Nombre secreto del tumor
Tipos de imagen
Modalidad de imagen
Nombre del tumor
Imagen de ejemplo
sí
Benigno o Maligno
región del cuerpo


Cirugía General,Traumatología y Ortopedia.
Los tumores óseos pueden afectar cualquier hueso del cuerpo y desarrollarse en cualquier parte del hueso, desde la superficie hasta el centro del hueso, llamada médula ósea. Un tumor óseo en crecimiento, incluso un tumor benigno, destruye el tejido sano y debilita el hueso, haciéndolo más vulnerable a las fracturas.
Cuando un tumor óseo es canceroso, es un cáncer óseo primario o un cáncer óseo secundario . Un cáncer de hueso primario en realidad comienza en el hueso, mientras que un cáncer de hueso secundario comienza en otra parte del cuerpo y luego hace metástasis o se disemina al hueso. El cáncer de hueso secundario también se llama enfermedad ósea metastásica.
Los tipos de cáncer que comienzan en otra parte y comúnmente se diseminan a los huesos incluyen:
Los cuatro tipos más comunes de cáncer de hueso primario son:
Hay muchos tipos de tumores óseos benignos, así como algunas enfermedades y afecciones que se asemejan a los tumores óseos. Aunque estas condiciones no son realmente tumores óseos, en muchos casos requieren el mismo tratamiento.
Algunos tipos comunes de tumores óseos benignos, y afecciones que comúnmente se agrupan con tumores, incluyen:
La mayoría de los tumores óseos, se desconoce la causa.
Los pacientes con un tumor óseo a menudo experimentan dolor en el área del tumor. Este dolor generalmente se describe como sordo y doloroso. Puede empeorar por la noche y aumentar con la actividad.
Otros síntomas de un tumor óseo pueden incluir fiebre y sudores nocturnos.
Muchos pacientes no tendrán ningún síntoma, pero en su lugar notarán una masa indolora.
Si bien los tumores óseos no son causados por un traumatismo, a veces una lesión puede hacer que un tumor comience a doler. La lesión también puede hacer que un hueso que está debilitado por un tumor se rompa o se rompa. Esto puede ser muy doloroso.
Ocasionalmente, los tumores benignos pueden descubrirse incidentalmente cuando se toma una radiografía por otro motivo, como un esguince de tobillo o una lesión en la rodilla.
Las infecciones, las fracturas por estrés y otras afecciones no tumorales pueden parecerse mucho a los tumores. Para asegurarse de que tiene un tumor óseo, su médico realizará una evaluación exhaustiva y ordenará una serie de pruebas.
Como parte del examen, su médico tomará un historial médico completo. Él o ella le preguntará sobre su salud general, los medicamentos que toma y sus síntomas actuales. Su médico también querrá saber si usted o algún miembro de su familia tiene antecedentes de algún tumor o cáncer.
Su médico realizará un examen físico completo, centrándose en la masa tumoral. Él o ella buscará:
En algunos casos, su médico examinará otras partes de su cuerpo para descartar cánceres que puedan propagarse a los huesos.
Rayos X. Los rayos X proporcionan imágenes de estructuras densas, como los huesos. En la mayoría de los casos, su médico ordenará una radiografía para ayudar a diagnosticar un tumor óseo. Los diferentes tipos de tumores pueden verse diferentes en las radiografías. Algunos disuelven el hueso o hacen un agujero en el hueso. Otros hacen que se forme hueso adicional. Algunos pueden hacer ambas cosas.
Otros estudios de imagen. Si es necesario, su médico ordenará una resonancia magnética nuclear (RMN), una tomografía computarizada (TC) o una gammagrafía ósea para ayudar a evaluar mejor su tumor.
Biopsia. Puede ser necesaria una biopsia para confirmar el diagnóstico de un tumor óseo. En una biopsia, se toma una muestra de tejido del tumor. Esta muestra es examinada bajo un microscopio y analizada por un patólogo (un médico que identifica enfermedades mediante el estudio de células anormales).
Existen dos métodos básicos para realizar una biopsia:
Otras pruebas. Su médico puede solicitar análisis de sangre y/u orina para ayudar a confirmar el diagnóstico de un tumor óseo o excluir otras afecciones.
Si su tumor es benigno, su médico puede recomendarle que lo controle de cerca para ver si cambia. Durante este tiempo, es posible que necesite radiografías de seguimiento periódicas u otras pruebas.
Algunos tumores benignos se pueden tratar eficazmente con medicamentos. Algunos desaparecerán con el tiempo sin cirugía.
Si tiene cáncer de huesos, el tratamiento incluirá un equipo de médicos de diferentes especialidades médicas que trabajarán juntos para brindar atención. Algunos serán oncólogos, médicos que se especializan en el tratamiento del cáncer. Su equipo puede incluir un oncólogo ortopédico, un oncólogo médico, un oncólogo radioterápico, un radiólogo y un patólogo. El objetivo del tratamiento es curar el cáncer mientras se mantiene la función, lo mejor posible, en la parte del cuerpo afectada por el tumor.
El tratamiento depende de varios factores, incluido el estadio del cáncer. Si el cáncer está localizado, las células cancerosas están contenidas en el tumor y el área circundante inmediata. Cuando el cáncer ha alcanzado una etapa metastásica, se ha diseminado a otras partes del cuerpo y puede ser más grave y más difícil de curar.
Los médicos a menudo combinan varios métodos para tratar los tumores óseos malignos:
Generalmente, los tumores malignos se extirpan mediante cirugía. A menudo, la radioterapia y la quimioterapia se usan en combinación con la cirugía.
En algunos casos, su médico puede recomendar la extirpación del tumor (escisión) u otra técnica quirúrgica para reducir el riesgo de fractura y discapacidad. Algunos tumores pueden reaparecer, incluso repetidamente, después del tratamiento adecuado. En raras ocasiones, ciertos tumores benignos pueden diseminarse o volverse cancerosos (hacer metástasis).
Cirugía de salvamento de extremidades. Esta cirugía extirpa la sección cancerosa del hueso, pero mantiene intactos los músculos, tendones, nervios y vasos sanguíneos cercanos en la medida de lo posible. El cirujano extraerá el tumor y una porción del tejido sano que lo rodea. El hueso extirpado se reemplaza con un implante metálico (prótesis), hueso de otra parte del cuerpo o hueso de un donante.
Amputación. La amputación es una cirugía para extirpar total o parcialmente un brazo o una pierna. Por lo general, se usa cuando un tumor es grande y/o los nervios y los vasos sanguíneos están involucrados. Una prótesis puede ayudar a la función después de la amputación.
La duración y la complejidad de su recuperación dependerá del tipo de tumor y del tipo de procedimiento que se haya realizado.
Cuando finaliza el tratamiento, su médico puede ordenar más radiografías y otros estudios de imágenes para confirmar que el tumor realmente desapareció.
Después del tratamiento, seguirá viendo a su médico para visitas de seguimiento regulares y pruebas cada pocos meses. Aunque el tumor haya desaparecido, es importante controlar su cuerpo para detectar signos de recurrencia. Los tumores que aparecen pueden plantear problemas graves, por lo que es importante detectarlos a tiempo.
Aunque tienen similitudes, los condromas periósticos y de tejidos blandos son menos comunes que los condromas que se forman en el interior del hueso (encondromas) y los que crecen fuera del hueso (osteocondromas).
Cuando se examinan a simple vista, los condromas aparecen como tumores brillantes de color blanco azulado con calcificaciones arenosas amarillas ocasionales. Bajo el microscopio, el tumor contiene cartílago azulado con una apariencia lobulada o dividida.
Los condromas periósticos también se conocen como condromas corticales, condromas yuxtacorticales, condromas pericorticales o condromas excéntricos. Aunque ocurren en personas de todas las edades, la mayoría de estos tumores ocurren en personas menores de 30 años.
Los condromas periósticos crecen lentamente durante meses o incluso años. Por lo general, miden de 1 a 2 cm de diámetro en los huesos tubulares de las manos y los pies, y hasta 3 o 4 cm en el húmero o el fémur.
En algunos casos, los condromas periósticos causarán hinchazón y un dolor sordo y doloroso. Algunas personas pueden sentir la masa tumoral, particularmente aquellas con condromas en los dedos de manos y pies. Los tumores en los dedos de las manos y los pies también tienen más probabilidades de causar dolor.
Con el tiempo, los condromas periósticos pueden erosionar el hueso subyacente y crear una depresión en forma de platillo. Se desarrolla un borde blanco duro (esclerosis) en el lado del tumor que mira hacia el hueso.
Los condromas de tejidos blandos se desarrollan más comúnmente durante la mediana edad. Aunque rara vez miden más de 3 cm de diámetro, a menudo se notan y aparecen como nódulos o bultos en los pequeños huesos de los dedos de las manos, las manos, los pies y los pies.
La mayoría de los pacientes tienen un solo tumor o masa que no duele.
Se desconoce la causa de los condromas periósticos y los condromas de tejidos blandos. No existen factores de riesgo conocidos (genética, lesión, infección o radiación) para estos tumores no cancerosos.
Antes de un examen físico, su médico hablará con usted sobre su salud general y sus síntomas para obtener un buen historial del problema. Durante el examen físico, su médico buscará sensibilidad sobre el hueso, hinchazón y una masa en el área de sus síntomas.
Su médico considerará otros tipos de masas tumorales durante el examen. Para diagnosticar con precisión cualquiera de los tipos de condroma, su médico ordenará pruebas de imagen o de tejido.
Rayos X. Estas pruebas brindan imágenes claras de estructuras densas como el hueso y son útiles para diagnosticar condromas.
En las radiografías, un condroma perióstico aparece como una sombra de tejido blando con erosión del hueso subyacente. A menudo están rodeados por un borde blanco (esclerótico). Un condroma perióstico siempre se desarrolla en el borde exterior del hueso. A veces, las células tumorales pueden calcificarse (comenzar a convertirse en hueso), lo que aparece como manchas blancas en una radiografía.
Los condromas de tejidos blandos no siempre aparecen en las radiografías porque no siempre se calcifican. Sin embargo, pueden causar erosiones óseas similares debido a un efecto de presión.
Otros escaneos de imágenes. Las tomografías computarizadas (TC) pueden ser útiles para evaluar el grado y las características del compromiso óseo. Además, una resonancia magnética nuclear (RMN) puede ayudar a definir mejor el tumor y su relación con el hueso. Estos escaneos pueden proporcionar más detalles, especialmente de los tejidos blandos. También pueden proporcionar imágenes transversales.
Biopsia. Si las pruebas de imagen no son concluyentes, su médico realizará una biopsia. En una biopsia, se toma una muestra de tejido del tumor y se examina bajo un microscopio. Su médico puede administrarle un anestésico local para adormecer el área y tomar una muestra con una aguja. Las biopsias también se pueden realizar como una operación pequeña.
Tanto los condromas periósticos como los condromas de tejidos blandos se identifican fácilmente como masas distintas y separadas y se pueden extirpar quirúrgicamente.
Procedimiento. Para extirpar completamente el tumor, su médico realizará un procedimiento quirúrgico llamado escisión. En este procedimiento, se le administrará un tipo de anestesia para adormecer el área alrededor del tumor. Después de administrar la anestesia, su médico hará una incisión en su piel y extirpará el tumor.
Reaparición. Estos tumores rara vez regresan después de la extirpación completa. No se necesita quimioterapia ni radiación.
El tiempo que lleve volver a las actividades diarias variará según el tamaño y la ubicación del tumor. La mayoría de los pacientes se recuperan rápidamente, excepto en la rara situación en la que se requiere una reconstrucción extensa de un hueso. En estos casos, la recuperación puede ser más larga y requerir inmovilización y uso limitado del área afectada. Su médico le dará instrucciones específicas para guiar su recuperación.
La mayoría de los condroblastomas se encuentran cerca de la articulación de la rodilla, ya sea en el extremo inferior del fémur (hueso del muslo) o en el extremo superior de la tibia (espinilla). También se encuentran en el hombro en la parte superior del húmero (hueso de la parte superior del brazo). Este tipo de condroblastoma a menudo se denomina “tumor de Codman”. Las ubicaciones menos comunes para los condroblastomas incluyen la pelvis, la cadera y el talón.
Los condroblastomas están formados por muchas células que se asemejan al cartílago fetal, de ahí su nombre (“condro” significa cartílago).
La mayoría de los huesos comienzan como cartílago. A medida que crece el feto, este cartílago se reemplaza principalmente por hueso. En los niños, los extremos de los huesos largos del cuerpo contienen una pequeña sección de cartílago llamada placa de crecimiento. Esta es el área del hueso donde ocurre el crecimiento. Cuando un niño está completamente desarrollado, las placas de crecimiento se cierran y se endurecen hasta convertirse en hueso sólido. En muchos casos, los condroblastomas se desarrollan alrededor del momento en que las placas de crecimiento comienzan a cerrarse.
Los hombres tienen el doble de probabilidades que las mujeres de desarrollar condroblastomas. El ochenta por ciento de los pacientes son menores de 25 años. Los tumores son muy raros y representan solo alrededor del 1 por ciento de todos los tumores óseos.
El condroblastoma es uno de los pocos tumores óseos benignos con el potencial de diseminarse o hacer metástasis a los pulmones. Aunque un condroblastoma más agresivo puede hacer metástasis, aún se considera un tumor benigno.
Se desconoce la causa de los condroblastomas. Se cree que los tumores surgen de las placas de crecimiento en los extremos de los huesos, cerca de las articulaciones. Sin embargo, los condroblastomas no producen el mismo tipo de cartílago normal que forma las placas de crecimiento o que rodea y protege las articulaciones.
No ha habido una conexión comprobada entre el desarrollo de los tumores y la exposición a sustancias químicas o radiación o cualquier actividad en particular.
El dolor es el síntoma más común de un condroblastoma. Debido a que los tumores generalmente se encuentran cerca de las articulaciones, es el dolor en las articulaciones lo que a menudo provocará una visita al médico. Los analgésicos de venta libre, como el paracetamol y el ibuprofeno, pueden ayudar a aliviar este dolor al principio, pero se volverán ineficaces a medida que el tumor crezca.
Otros signos y síntomas de un condroblastoma pueden incluir:
Un condroblastoma suele ser pequeño y está contenido dentro del hueso, por lo que los pacientes normalmente no ven ni sienten una masa.
Su médico hablará con usted sobre su historial médico y salud general y le preguntará acerca de sus síntomas.
Él o ella tratará de aprender tanto como sea posible acerca de su dolor. El dolor por la noche o el dolor que no desaparece con el descanso suele ser más preocupante. Esto se debe a que el dolor causado por una lesión generalmente disminuirá cuando no se use el área lesionada.Luego, su médico examinará el área adolorida en busca de:
Su médico usará estudios por imágenes y otras pruebas para ayudar a diagnosticar un condroblastoma.
Rayos X. Estos estudios proporcionan imágenes de estructuras densas, como el hueso, y son muy útiles para diagnosticar condroblastomas.
La mayoría de los condroblastomas son tumores redondos pequeños (de 1 a 4 cm). En las imágenes de rayos X, a menudo están rodeados por un borde delgado de hueso blanco. Algunos harán que el borde del hueso se salga, pero rara vez los tumores se extienden más allá del hueso y hacia el tejido blando circundante. En alrededor del 25 al 40 por ciento de los casos, una radiografía mostrará calcificaciones (manchas blancas) dentro del tumor.
 La radiografía muestra un condroblastoma en el extremo superior del húmero (hueso de la parte superior del brazo). Tenga en cuenta los bordes blancos que delimitan el tumor. (Derecha) Una resonancia magnética del mismo tumor.")
Exploraciones de tomografía computarizada (TC). Más detallada que una radiografía simple, una tomografía computarizada puede ayudar a su médico a evaluar más a fondo el tumor y planificar su tratamiento. Es más probable que las calcificaciones dentro del tumor sean visibles en una tomografía computarizada.
Imágenes por resonancia magnética (IRM). Estos estudios proporcionan imágenes claras de los tejidos blandos del cuerpo. Una resonancia magnética ayudará a su médico a ver mejor los bordes del tumor y determinar si se ha expandido fuera del hueso y hacia los tejidos cercanos. También mostrará áreas de inflamación que normalmente rodean un tumor.

Biopsia. A menudo es necesaria una biopsia para confirmar el diagnóstico de condroblastoma. En una biopsia, se toma una muestra de tejido del tumor y se examina bajo un microscopio.
Una biopsia se puede realizar bajo anestesia local con una aguja o como una pequeña operación abierta.
Bajo un microscopio, los condroblastomas tienen un fondo que parece cartílago y una mezcla de células, algunas de las cuales parecen células productoras de cartílago (éstas tienen núcleos que parecen granos de café). Las calcificaciones se pueden ver en todo el tumor en un patrón que se asemeja a “alambre de gallinero”.
Las pruebas ayudarán a su médico a diferenciar un condroblastoma de otros tumores que tienen una apariencia similar, como un tumor de células gigantes, un encondroma, un quiste óseo aneurismático, una infección del hueso (osteomielitis) o un condrosarcoma de células claras.
Es muy importante diferenciar un condroblastoma de un tumor más agresivo como un condrosarcoma. Un patólogo (un médico que identifica enfermedades mediante el estudio de células anormales) revisará la muestra de tejido y realizará pruebas especiales para ayudar a determinar el diagnóstico.
Sin tratamiento, un condroblastoma continuará creciendo y destruyendo el hueso circundante, por lo que siempre es necesario el tratamiento. Los objetivos del tratamiento son:
La cirugía es siempre el tratamiento preferido para los condroblastomas; sin embargo, hay algunos casos en los que un tumor no se puede extirpar de manera segura o efectiva debido a su ubicación o tamaño. En este caso, su médico puede recomendar un tratamiento no quirúrgico.
El tratamiento no quirúrgico puede incluir:
Ablación por radiofrecuencia. En este procedimiento, el tumor se calienta y se destruye con una corriente eléctrica de alta frecuencia.
Crioterapia. En este procedimiento, el tumor se destruye usando frío extremo producido por nitrógeno líquido.
Tanto la ablación por radiofrecuencia como la crioterapia se utilizan normalmente cuando la cirugía puede provocar complicaciones inaceptables.
Siempre que sea posible, se utiliza la cirugía para tratar los condroblastomas. El tratamiento quirúrgico puede incluir:
Legrado. Este es el procedimiento más comúnmente utilizado para tratar un condroblastoma. En el legrado, se utilizan instrumentos especiales para raspar el tumor del hueso. Una vez que se le cure un condroblastoma, es inusual que regrese.
Injerto óseo. Después del legrado, su médico puede llenar la cavidad con un injerto óseo para ayudar a estabilizar el hueso. Un injerto óseo se obtiene de un donante (aloinjerto) o de otro hueso del cuerpo (autoinjerto), generalmente de la pelvis.
Su médico también puede usar una mezcla de cemento óseo para rellenar el orificio. A veces, se colocan productos químicos adicionales, como fenol o nitrógeno líquido, dentro de la cavidad ósea para tratar de reducir el riesgo de recurrencia.

Resección. Según la ubicación y el tamaño del tumor, su médico puede extirpar toda la sección de hueso que contiene el tumor, en lugar de realizar un legrado. Luego se pueden usar barras o placas de metal y tornillos para estabilizar el hueso.
En casos raros, un condroblastoma puede diseminarse a los pulmones u otros órganos. Si el tumor se propaga, es necesaria la extirpación quirúrgica del hueso, así como del área afectada del órgano. Esto típicamente resultará en una cura.
La recurrencia del tumor es la complicación más grave del tratamiento quirúrgico. Otras posibles complicaciones incluyen:
Su resultado después del tratamiento dependerá de una serie de factores, que incluyen:
Incluso con una cirugía exitosa, los condroblastomas pueden dañar el cartílago normal que rodea y protege las articulaciones, por lo que algunos pacientes pueden desarrollar artritis con el tiempo.
Los condroblastomas recurren en aproximadamente el 10 por ciento de los pacientes. Si un tumor reaparece, por lo general lo hace dentro de unos pocos meses a unos pocos años. Por este motivo, su médico le hará un seguimiento con controles periódicos y radiografías durante al menos unos años. Si un tumor reaparece, se puede tratar con los mismos métodos. Su médico hablará con usted sobre todas sus opciones.
El fibroma condromixoide crece a partir del tejido formador de cartílago que se encuentra en la médula de los huesos (“condro” significa cartílago). Por lo general, se desarrolla en el extremo ensanchado de un hueso largo en la parte inferior del cuerpo, como la tibia (espinilla) o el fémur (hueso del muslo). La ubicación más común de CMF es en la tibia cerca de la rodilla. Otras ubicaciones comunes incluyen el fémur cerca de la rodilla, los huesos del pie y la pelvis (área de la cadera).
CMF no hace metástasis (diseminación) a otras partes del cuerpo.
Se desconoce la causa de la CMF. No ha habido una conexión comprobada entre el desarrollo de CMF y la exposición a productos químicos, radiación o cualquier actividad en particular.
Las personas con fibroma condromixoide pueden sentir un bulto en la ubicación del tumor. Puede haber un dolor de leve a severo asociado con la protuberancia, o puede no haber dolor en absoluto. En algunos casos, no hay protuberancia, solo una cantidad variable de dolor en el área del tumor.
Hay muchos aspectos en el examen del médico. Antes de un examen físico, su médico hablará con usted sobre su estado de salud general, así como sobre sus síntomas, para obtener un buen historial del problema. El dolor nocturno o en reposo, o el dolor que simplemente no desaparece, es más típico de los tumores en general y del CMF en particular. Por el contrario, el dolor de una lesión generalmente disminuye cuando el área lesionada no se mueve.
Durante el examen físico, su médico buscará sensibilidad sobre el hueso y verificará su rango de movimiento en el área de su dolor.
Para diagnosticar CMF, su médico ordenará pruebas de imagen y de tejido.
Rayos X. Estas pruebas brinda imágenes claras de estructuras densas como el hueso y son muy útiles para diagnosticar la CMF. La mayoría de los fibromas condromixoides son tumores pequeños (de 1 a 4 cm), de forma redonda u ovalada. En las radiografías, a menudo están rodeados por un borde blanco (esclerótico).
En muchos casos, el tumor destruirá porciones del hueso y crecerá agresivamente, empujando hacia los tejidos blandos circundantes. Es por eso que algunos pacientes pueden sentir un bulto a través de la piel.
Otros escaneos de imágenes. Por lo general, los médicos también solicitan tomografías computarizadas (TC) o imágenes por resonancia magnética (IRM) para ayudar a definir mejor el tumor. Estos escaneos pueden proporcionar más detalles, especialmente de los tejidos blandos. También pueden proporcionar imágenes transversales.
Biopsia. A menudo es necesaria una biopsia para confirmar el diagnóstico de CMF. En una biopsia, se toma una muestra de tejido del tumor y se examina bajo un microscopio. Su médico puede administrarle un anestésico local para adormecer el área y tomar una muestra con una aguja. Las biopsias también se pueden realizar como una operación pequeña.
Bajo un microscopio, los fibromas condromixoides tienen elementos de cartílago benigno (condroide), mixoide y tejido fibroso.
Las imágenes y las pruebas de tejido ayudan a su médico a diferenciar el CMF de otros tumores que tienen una apariencia similar, como el tumor de células gigantes, el encondroma, el condroma, el fibroma no osificante, el quiste óseo aneurismático o la infección del hueso (osteomielitis).
Al planificar el tratamiento, es muy importante diferenciar el CMF de tumores cancerosos más agresivos como el condrosarcoma o el osteosarcoma.
El tratamiento de los fibromas condromixoides requiere cirugía.
El método más común para tratar la CMF es el curetaje. En este procedimiento, se raspa el tumor del hueso.
Después del legrado, su médico puede llenar el orificio con un injerto óseo; se trata de hueso tomado de un donante (aloinjerto) o de otro hueso de su cuerpo (autoinjerto). A veces, se colocan químicos adicionales, como fenol o nitrógeno líquido, dentro de la cavidad ósea para tratar de reducir el riesgo de que el tumor vuelva a crecer.
La recurrencia del tumor es la complicación más grave del tratamiento. Incluso cuando estos tumores se tratan adecuadamente, vuelven a crecer en el mismo lugar hasta en un 25 % de las veces. Una cirugía más agresiva, como la extirpación de porciones más grandes del hueso, disminuirá la posibilidad de que el tumor regrese, pero puede tener una mayor probabilidad de causar complicaciones o dañar el hueso de forma permanente.
Otras complicaciones incluyen infección y, en raras ocasiones, fractura del hueso a través del área tratada.
El tiempo que le llevará regresar a todas sus actividades diarias variará según la ubicación del tumor. Si tiene algún dolor o molestia, es posible que desee limitar alguna actividad. Su médico le dará instrucciones específicas para guiar su recuperación.
Su médico lo controlará y le tomará radiografías de rutina durante algunos años después de la cirugía para detectar la recurrencia del tumor. Si el tumor regresa, se puede tratar con los mismos métodos. Su médico le hablará sobre todas las opciones.
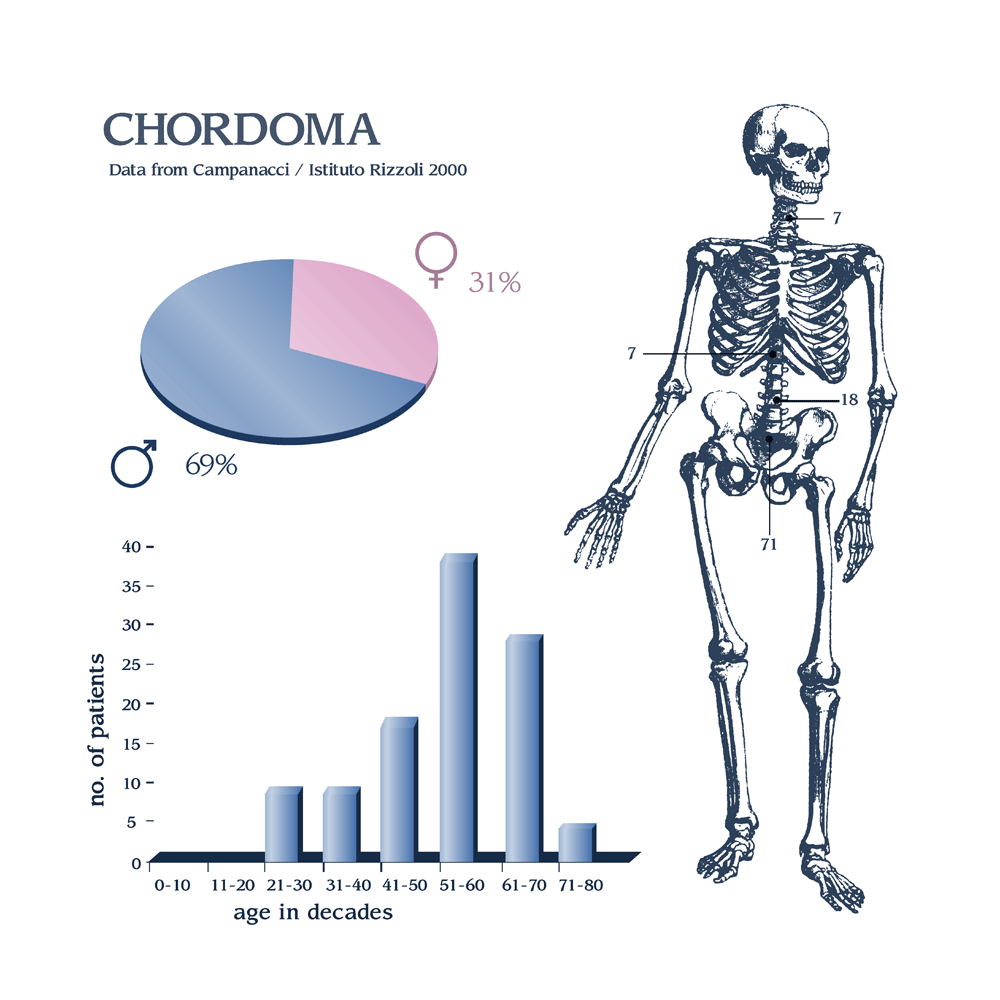
Los cordomas se desarrollan en las células sobrantes de la notocorda , una estructura similar al cartílago que ayuda a formar la columna vertebral temprana en las etapas iniciales del desarrollo fetal. La mayor parte de la notocorda es reemplazada por la columna vertebral durante los primeros seis meses de desarrollo. Sin embargo, pueden quedar pequeñas áreas y se cree que se pueden formar cordomas en estas áreas.
Los cordomas pueden ocurrir en cualquier lugar a lo largo de la columna, pero con mayor frecuencia ocurren en la base de la columna (sacro), en el coxis (coxis) o en la base del cráneo.

La mayoría de los cordomas ocurren en pacientes entre 40 y 70 años de edad. Ocasionalmente, ocurren en pacientes más jóvenes, incluidos los niños.
Los cordomas son tumores potencialmente mortales. En algunos casos, pueden extenderse o hacer metástasis a otras áreas del cuerpo. Cuando los cordomas se propagan, con mayor frecuencia se propagan a los pulmones.
Los cordomas ocurren espontáneamente. No se sabe que sean causados por traumatismos, factores ambientales o dieta. Los cordomas no se heredan. No están asociados con ninguna condición médica ni con el uso de ningún medicamento o suplemento.
Debido a que los cordomas crecen lentamente, los pacientes a menudo tienen síntomas durante mucho tiempo, en algunos casos, más de un año, antes de buscar atención médica.
Los síntomas dependen de la ubicación del tumor:
Su médico realizará un examen físico completo y utilizará estudios de imágenes para diagnosticar un cordoma.
Es posible que se necesiten varios estudios de imágenes para identificar el tumor. Estos pueden incluir:
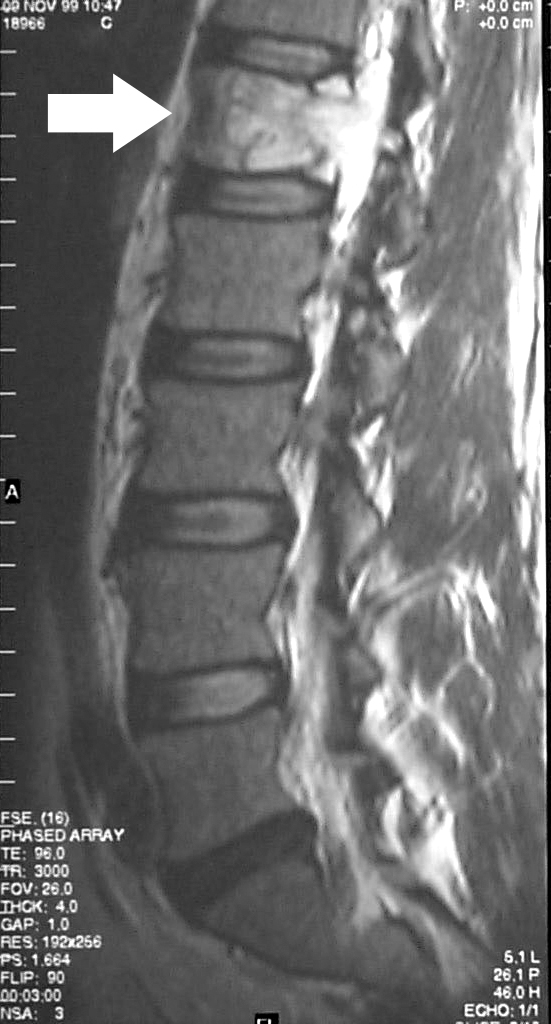
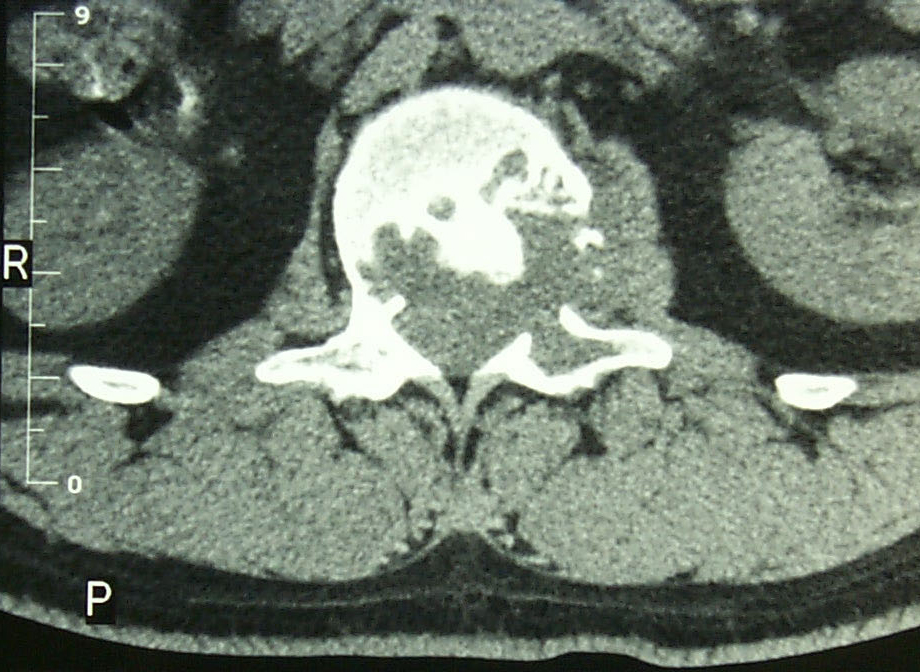
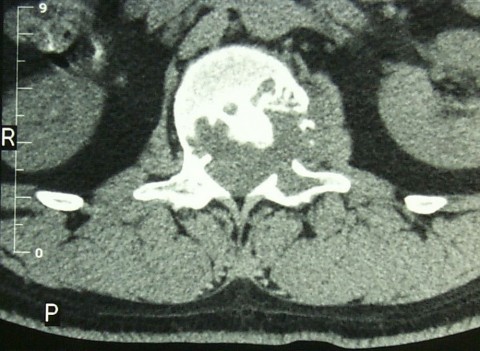
Si bien los cordomas en sí mismos no se ven bien en las radiografías, una radiografía puede revelar daño óseo causado por el tumor. Las radiografías del sacro suelen ser difíciles de interpretar para los médicos.
Una resonancia magnética se considera la mejor prueba para determinar el tamaño total del tumor.
Una biopsia es necesaria para confirmar un diagnóstico de cordoma. En una biopsia, se toma una muestra de tejido del tumor y se examina bajo un microscopio.
Una vez que el tumor haya sido identificado como cordoma, su médico realizará más pruebas para determinar si el cáncer se ha propagado. Este proceso se conoce como “puesta en escena”. Estas pruebas adicionales pueden incluir:
La parte del cuerpo donde se desarrolla el primer tumor se denomina sitio “primario”. Cualquier parte del cuerpo donde se haya diseminado se llama sitio “metastásico”.
Al identificar la etapa del tumor, su médico puede determinar la estrategia de tratamiento más efectiva.
Debido a su proximidad con el cerebro y la médula espinal, los cordomas son muy difíciles de tratar.
La quimioterapia tradicional no suele ser muy eficaz y la radiación estándar por sí sola rara vez cura el tumor.
Siempre que sea posible, la cirugía es la opción de tratamiento preferida. La cirugía a menudo se combina con radiación para tratar los cordomas. Si un tumor no se puede extirpar quirúrgicamente, la radioterapia puede ayudar a detener el crecimiento del tumor. Las técnicas de radiación más nuevas, como la terapia con haz de protones, pueden ser más eficaces que la radiación estándar para detener el crecimiento del tumor.
Aunque es la opción de tratamiento preferida, la cirugía a menudo puede ser difícil de realizar debido a las estructuras importantes que se encuentran cerca del tumor. Para garantizar el mejor resultado, el cirujano debe extirpar el tumor y una parte del tejido normal que lo rodea. Esto a menudo resulta en alguna pérdida de la función neurológica. Por ejemplo, la extirpación de tumores en el sacro puede provocar la pérdida del control de los intestinos y la vejiga (incontinencia), debilidad en las extremidades inferiores o pérdida de sensibilidad en el área de la ingle. También puede causar inestabilidad de los huesos pélvicos. Su médico hablará con usted sobre los riesgos en su caso específico.
Su resultado después del tratamiento depende de una serie de factores, que incluyen:
Su cirujano hablará con usted sobre su pronóstico individual.
Los avances en la radioterapia han permitido que los pacientes reciban dosis más altas de radiación con menos daño a los tejidos normales circundantes, como el cerebro y la médula espinal. Los diferentes tipos de radioterapia (incluida la terapia con haz de protones, la radiación intraoperatoria y la braquiterapia) pueden ofrecer un mejor tratamiento de los cordomas. Estas radioterapias pueden combinarse con una cirugía más limitada en el futuro.
Actualmente se están investigando nuevos tratamientos farmacológicos, incluido el uso de un fármaco llamado mesilato de imatinib. Estos medicamentos pueden ayudar a retrasar el crecimiento de tumores que no se pueden extirpar con cirugía.
Los encondromas pueden ocurrir en cualquier persona, pero son más comunes en pacientes de entre 10 y 20 años. Se encuentran con mayor frecuencia en los huesos pequeños de la mano. De hecho, el encondroma es el tumor más común en la mano. Los encondomas también pueden desarrollarse en los huesos largos del cuerpo, como el fémur (hueso del muslo), la tibia (hueso de la espinilla) y el húmero (hueso de la parte superior del brazo).
Los encondromas suelen ser tumores solitarios. En casos raros, sin embargo, múltiples tumores pueden aparecer como parte de una condición como la enfermedad de Ollier o el síndrome de Maffucci.
Los encondromas únicos rara vez se vuelven cancerosos, aunque las posibilidades son un poco más altas en pacientes con la enfermedad de Ollier y el síndrome de Maffucci. Cuando los encondromas se vuelven cancerosos, generalmente se convierten en un tipo de tumor de cartílago maligno llamado condrosarcoma .
Distinguir entre un encondroma no canceroso y la forma de muy bajo grado de un tumor canceroso puede ser difícil, incluso para los cirujanos ortopédicos de tumores.
Se desconoce la causa exacta de los encondromas. Algunas investigaciones indican que pueden resultar cuando las células se convierten en cartílago en lugar de hueso.
No se cree que los tumores sean causados por radiación o exposición química o por alguna actividad específica.
En la mayoría de los casos, los encondromas no son dolorosos y no causan ningún síntoma. Sin embargo, si los tumores aparecen en manos o pies, o si hay múltiples lesiones, el hueso puede debilitarse y deformarse. Esto puede provocar fracturas óseas patológicas y agrandamiento de los dedos afectados.
En pacientes con enfermedad de Ollier y síndrome de Maffucci, las deformidades óseas pueden ser bastante graves. Si se ha excluido el dolor de otras fuentes, su médico estudiará cuidadosamente el tumor para determinar si en realidad podría ser un condrosarcoma de bajo grado. El dolor durante la noche o en reposo es más probable que indique un tumor maligno. Sin embargo, debido a que el dolor es un síntoma común de muchas afecciones y lesiones, su médico realizará una evaluación exhaustiva.
Debido a que no suelen causar síntomas, la mayoría de los encondromas se detectan cuando se toman radiografías de rutina por otro motivo, como una lesión o artritis. Cuando esto ocurra, su médico realizará un examen y ordenará una serie de pruebas para confirmar que su tumor es en realidad un encondroma, y no un tumor más agresivo o canceroso.
Durante el examen, su médico tomará un historial médico completo y le preguntará acerca de sus síntomas. Él o ella le preguntará si su tumor es doloroso y cuándo ocurre el dolor. Existe una mayor preocupación si el dolor ocurre cuando está en reposo o durante la noche y no desaparece. El dolor causado por la actividad es menos preocupante.
En algunos casos, su médico puede administrarle una inyección en la articulación cercana al tumor. Si la inyección alivia su dolor, indica que el encondroma no es la causa.
Rayos X. Los rayos X proporcionan imágenes de estructuras densas como los huesos. En las radiografías, los encondromas aparecen como tumores pequeños (menos de 5 cm), con forma de lóbulo, oscurecidos en el medio del hueso. Suelen contener manchas blancas o calcificación en su interior. Las áreas blancas del tumor muestran un patrón de anillos y arcos que indica que el tumor contiene cartílago.
Otros estudios de imagen. Su médico puede solicitar una tomografía computarizada (TC) o una resonancia magnética nuclear (RMN) para ayudar a evaluar mejor su tumor. Estas exploraciones brindan una imagen más completa del hueso que rodea el tumor. Si el tumor se ha vuelto maligno, las exploraciones pueden mostrar erosión ósea, inflamación ósea o una masa que crece fuera del hueso.
En algunos casos, su médico puede ordenar una gammagrafía ósea. Durante esta prueba, se inyecta una cantidad muy pequeña de colorante radiactivo en el cuerpo por vía intravenosa. Tanto los tumores benignos como los malignos pueden provocar un aumento de la absorción del material radiactivo en el hueso debido a la actividad ósea. Los encondromas suelen estar activos en las gammagrafías óseas.
Biopsia. Puede ser necesaria una biopsia para confirmar el diagnóstico de encondroma. En una biopsia, se toma una muestra de tejido del tumor y se examina bajo un microscopio.
Una biopsia se puede realizar bajo anestesia local con una aguja o como una pequeña operación abierta.
Calificación
El grado, o la agresividad, del tumor se determina mediante estudios de imágenes y cómo se ve el tumor bajo un microscopio.
Bajo el microscopio, los encondromas tienen islas de cartílago que son fáciles de distinguir del hueso normal que las rodea. Por lo general, el cartílago no se encuentra en el centro de los huesos. Los encondromas en la mano y el pie o en pacientes con la enfermedad de Ollier o el síndrome de Maffucci pueden contener cartílago de aspecto más extraño. Puede ser difícil distinguir estos tumores de los condrosarcomas de bajo grado.
Los condrosarcomas de bajo grado se ven más celulares que los encondromas bajo un microscopio y hay menos hueso normal en el tumor. Debido a que los condrosarcomas y los encondromas de bajo grado se ven similares, los cirujanos, radiólogos y patólogos experimentados trabajarán juntos para obtener la mejor interpretación del tumor.
Las características de un tumor más agresivo o un condrosarcoma maligno incluyen:
Si su tumor no causa síntomas, su médico puede recomendar observación y control para ver si crece. Durante este tiempo, es posible que necesite radiografías periódicas u otras pruebas. La mayoría de los médicos creen que no es necesario extirpar los tumores sin síntomas.
El curetaje es el procedimiento quirúrgico más utilizado para tratar los encondromas. En el legrado, se raspa el tumor del hueso. Una vez que se eliminan los encondromas, la mayoría no volverá. Si un tumor ha causado la fractura de su hueso, su médico generalmente permitirá que la fractura sane antes de tratar el tumor. Luego, se le raspará el tumor para evitar otra fractura.
Después del legrado, su médico puede llenar la cavidad con un injerto óseo para estabilizar el hueso. Un injerto óseo es hueso extraído de un donante (aloinjerto) o de otro hueso del cuerpo (autoinjerto). En algunos casos, se puede usar otra sustancia para llenar la cavidad.
Algunos tumores pueden parecer simples encondromas en las radiografías, pero son dolorosos. El tratamiento de estas lesiones puede ser controvertido. Algunos médicos recomiendan el legrado quirúrgico. Otros piensan que no es probable que los tumores sean la causa del dolor en el área, por lo que recomiendan monitorear con radiografías periódicas.
Desafortunadamente, las biopsias no suelen ser útiles en estos casos. Incluso para patólogos especializados en huesos, puede ser difícil diferenciar entre un encondroma benigno y un condrosarcoma de bajo grado. En este contexto, no se recomiendan las biopsias con aguja.
Los tumores más agresivos con destrucción ósea o con una masa que crece fuera del hueso suelen ser condrosarcomas. Estos tumores malignos deben extirparse en su totalidad. El procedimiento específico utilizado depende del grado del tumor.
Hay una gran cantidad de investigaciones en curso sobre encondromas y condrosarcomas. Los estudios actuales están tratando de identificar marcadores químicos que puedan ayudar a los médicos a diferenciar entre tumores de cartílago benignos y malignos.
Otros estudios están analizando las diferentes características de los tumores en estudios de imágenes especializados, como resonancias magnéticas, tomografías computarizadas o tomografías por emisión de positrones (PET).
En comparación con otros tipos de cáncer, los tumores óseos malignos como el sarcoma de Ewing son raros. De estos raros tumores óseos, el sarcoma de Ewing es el segundo más común en niños y adultos jóvenes. Según datos de niños menores de 15 años, aproximadamente 1,7 de cada millón de niños desarrollan la enfermedad.
Recientemente, los médicos han definido la enfermedad para incluir cuatro tipos de cáncer, denominados la Familia de Tumores de Ewing (EFT). Esto incluye el sarcoma óseo de Ewing, el sarcoma de tejido blando de Ewing, el tumor neuroectodérmico primitivo (PNET) que puede ocurrir tanto en el hueso como en el tejido blando, y el tumor de Askin, un PNET que ocurre en los huesos del tórax.
Este artículo se centra en el sarcoma óseo de Ewing.
No se conoce la causa del sarcoma de Ewing.
Se sabe que la mayoría de los cánceres surgen de cierto tipo de tejido u órgano. Por ejemplo, el cáncer de mama surge de las células mamarias. Sin embargo, en el caso del sarcoma de Ewing, los médicos no conocen el tipo exacto de célula donde se origina el cáncer.
Lo que se sabe es que el cáncer se forma cuando ocurren cambios en los cromosomas de una célula. En las células del sarcoma de Ewing, el material genético de los cromosomas n.° 11 y n.° 22 no coincide. Esta anomalía genética no se hereda de los padres del niño; los cambios cromosómicos ocurren después del nacimiento del niño.
No se entiende por qué ocurre esta anomalía. Los médicos no han identificado ningún factor de riesgo que haga que un niño sea más susceptible que otro. El tumor no se desarrolla como resultado de ningún hábito dietético, social o conductual. No se conocen formas de prevenir la enfermedad, y los padres deben saber que no hay nada que pudieran haber hecho diferente para prevenir el tumor de su hijo.
Por lo general, hay dolor y posiblemente hinchazón en el sitio del tumor. Sin embargo, el tumor puede estar presente durante muchos meses antes de que crezca lo suficiente como para causar dolor e inflamación. En algunos casos, el primer síntoma del sarcoma de Ewing es la presencia de una masa.
Aunque las lesiones no son una causa conocida, una lesión puede llamar la atención sobre un tumor. Por ejemplo, un hueso debilitado por un tumor puede romperse después de una lesión menor.
Después de analizar los síntomas y realizar un examen físico, su médico usará varios tipos de pruebas para diagnosticar el sarcoma de Ewing.
Primero, un médico usa estudios de imágenes para crear imágenes detalladas del área afectada. Estos incluyen radiografías, imágenes por resonancia magnética (IRM), tomografías computarizadas (TC) y gammagrafías óseas.
Para confirmar que el tumor observado en los estudios por imágenes es un sarcoma de Ewing, su médico realizará una biopsia. Esto implica tomar un trozo de tejido del tumor y observarlas bajo un microscopio. Una biopsia es un procedimiento simple que se puede realizar en un quirófano o en un departamento de rayos X.
Los tumores del sarcoma de Ewing a veces se denominan tumores de células azules pequeñas (células redondas) debido a la forma en que se ven bajo el microscopio.
Una vez que el tumor se identifique como sarcoma de Ewing, su médico realizará más pruebas para determinar si el cáncer se ha propagado. Este proceso se conoce como “puesta en escena”. Estas pruebas adicionales pueden incluir:
La parte del cuerpo donde se desarrolla el primer tumor se denomina sitio “primario”. Cualquier parte del cuerpo donde se haya diseminado se llama sitio “metastásico”.
Al identificar la etapa del tumor, su médico puede determinar la estrategia de tratamiento más efectiva.
En el momento del diagnóstico, muchos casos de sarcoma de Ewing no se han propagado a otras partes del cuerpo. Sin embargo, incluso si las pruebas no muestran propagación, los médicos planificaron una estrategia de tratamiento que asume que ya se ha producido una cantidad muy pequeña de propagación (enfermedad micro metastásica).
Los médicos de muchas especialidades ayudan a tratar el sarcoma de Ewing. Estos incluyen oncólogos quirúrgicos ortopédicos, oncólogos médicos pediátricos o de adultos, oncólogos radioterápicos, patólogos y radiólogos. La mayoría de los pacientes son tratados en las principales instituciones hospitalarias o centros oncológicos.
Los principales tratamientos son la quimioterapia, la cirugía y la radiación. Estos tratamientos a menudo se usan en alguna combinación entre sí.
Tanto la cirugía como la radiación son tratamientos efectivos para extirpar el tumor primario. En la mayoría de los casos, los médicos recurren a la cirugía para extirpar los tumores cuando es posible. El tratamiento de radiación se usa solo cuando la cirugía no puede extirpar completamente el tumor o haría que el paciente pierda la función en el área afectada.
La mayoría de los médicos trabajan en equipos para adaptar sus recomendaciones de cirugía y/o radiación a la situación específica de un paciente.
La quimioterapia es un tratamiento farmacológico que se usa para destruir el tumor primario y cualquier diseminación del cáncer. A menudo se usa inicialmente para reducir el tamaño del tumor primario y facilitar su extirpación con cirugía.
Debido a que el tratamiento de quimioterapia para el sarcoma de Ewing dura mucho tiempo, los medicamentos se administran a través de un catéter venoso central permanente. Este es un tubo muy delgado que se inserta en una vena en el pecho y permanecerá en su lugar hasta el final del tratamiento. Se coloca justo antes de iniciar el tratamiento de quimioterapia.
La quimioterapia se realiza en ciclos. Utiliza combinaciones de varios medicamentos para destruir las células cancerosas. Debido a que estos medicamentos afectan todo el sistema, las células sanas también pueden dañarse, incluidos los glóbulos blancos y las plaquetas. El período de descanso entre ciclos permite que se recupere el conteo de células sanguíneas, pero todavía hay efectos secundarios por el daño celular. Los efectos secundarios más comunes incluyen pérdida de cabello, náuseas, llagas en la boca y fiebre. Ciertos medicamentos pueden ayudar a aliviar algunos efectos secundarios.
La cirugía o la radiación para extirpar el tumor primario se pueden realizar antes o durante la quimioterapia. La mayoría de las veces, esto ocurre después de varios ciclos de quimioterapia. Después de la recuperación de la cirugía y/o la radioterapia, se reanudará la quimioterapia. Generalmente toma cerca de un año terminar todos los ciclos de quimioterapia.
Se utilizan varios medicamentos en el tratamiento de quimioterapia para el sarcoma de Ewing. Los fármacos más comunes son la vincristina, la dactinomicina, la ciclofosfamida y la doxorrubicina. La evidencia reciente muestra que la adición de ifosfamida y etopósido ayuda.
Se han logrado muchos avances en la quimioterapia a través del conocimiento adquirido al colocar a los pacientes en ensayos clínicos. Los médicos solicitan permiso para inscribir a los pacientes en ensayos clínicos específicos. Su médico puede brindarle más información sobre cualquier ensayo clínico que se esté realizando actualmente.
La cirugía se utiliza para extirpar el tumor primario y, en ocasiones, cualquier tumor más pequeño, si la enfermedad se ha propagado. El tumor debe extirparse por completo para evitar que regrese, y también se extirpa un margen de músculo sano y tejido articular que rodea el tumor.
Los cirujanos pueden reconstruir el hueso, la articulación o el sitio del tejido blando. Pueden usar injertos óseos, articulaciones artificiales o una combinación de estos. El objetivo es restaurar la parte del cuerpo para que el paciente pueda realizar sus actividades cotidianas normales.
La cantidad de función que se puede restaurar a partir de la cirugía reconstructiva depende de la cantidad de tejido muscular y articular que se pueda preservar mientras se extirpa todo el tumor.
Según el sitio de la cirugía, es posible que el paciente deba limitar el peso sobre la extremidad reconstruida. La rehabilitación continua con fisioterapia es necesaria para volver a las actividades diarias. Un fisioterapeuta puede proporcionar ejercicios para ayudar a mantener el rango de movimiento y recuperar la fuerza.
Es probable que las actividades atléticas o extenuantes causen demasiado estrés o desgaste en la reconstrucción, pero ocasionalmente algunos pacientes aún intentarán realizar estas actividades. Sin embargo, la mayoría de los pacientes no podrán realizar dichas actividades debido a la cantidad de tejido muscular y articular extirpado.
Algunos pacientes pueden necesitar más operaciones para mantener la extremidad en funcionamiento por el resto de sus vidas. La reconstrucción de un hueso en un niño en crecimiento es un desafío especial. A medida que el niño crece, se necesitan múltiples procedimientos para alargar el hueso reconstruido.
Es importante considerar los riesgos y complicaciones asociados con la cirugía. Las infecciones, los problemas con la articulación artificial y la cicatrización de heridas son las preocupaciones más frecuentes.
El objetivo principal de la cirugía tumoral es extirpar el tumor por completo y evitar que regrese; los temas reconstructivos, aunque importantes, son sin embargo un objetivo secundario.
Algunos tumores de sarcoma de Ewing no se pueden extirpar de manera segura con cirugía. En estos casos, los médicos suelen utilizar radioterapia para destruir el tumor. Ocasionalmente, un tumor requiere la amputación de la extremidad para garantizar que el tumor y los tejidos circundantes se hayan extirpado por completo.
La radioterapia puede destruir las células cancerosas y reducir los tumores. Se usa con más frecuencia para reducir la posibilidad de que el tumor primario regrese (recurrencia). La radiación también se puede usar en lugar de la cirugía en sitios donde la cirugía es demasiado riesgosa o complicada. Ocasionalmente, la radioterapia se usa junto con la cirugía.
Cuando se utiliza el tratamiento con radiación, se administran tratamientos diarios durante el transcurso de varias semanas. Si bien se puede evitar la incomodidad de la cirugía, existen riesgos asociados con la radiación, que incluyen:
Con los años, el resultado de los pacientes con sarcoma de Ewing ha mejorado considerablemente. Esto es gracias a las mejoras en la quimioterapia, el diagnóstico por imágenes y las técnicas reconstructivas.
El pronóstico varía de un paciente a otro pero, en general, dos tercios de los pacientes sin propagación de la enfermedad sobrevivirán al menos cinco años después del diagnóstico con el tratamiento estándar.
Varios factores pueden afectar la probabilidad de supervivencia. Éstas incluyen:
Los sobrevivientes del sarcoma de Ewing requieren atención de seguimiento continua para monitorear cualquier efecto secundario tardío del tratamiento y cualquier recurrencia del tumor. Cuando los tumores regresan, generalmente ocurre dentro de los primeros años después del tratamiento.
El tratamiento para el sarcoma de Ewing cambiará en los próximos años a medida que se disponga de nuevos conocimientos. La investigación actual sobre la familia de tumores de Ewing se ha centrado en muchas áreas.
No se conoce el significado de la anomalía cromosómica, ni se sabe cómo afecta esta a la función celular normal. El conocimiento básico en esta área podría conducir algún día a nuevas terapias que puedan aprovechar las anomalías en la función celular. Puede dirigirse a las células cancerosas.
Otras investigaciones involucran quimioterapia y abordan cuestiones sobre la dosis adecuada de medicamentos y las mejores combinaciones de medicamentos. Los médicos también están analizando varios tipos de trasplantes de médula ósea.
Los ingenieros biomecánicos y los cirujanos siempre realizan mejoras en las articulaciones artificiales. Las prótesis expandibles, que se pueden alargar a medida que el niño crece, ya están disponibles.
Los médicos están investigando los factores de crecimiento tisular. Podrían ayudar a curar huesos rotos.
Tumores desmoides extra abdominales
Los tumores desmoides son fibrosos, muy parecidos al tejido cicatricial. Por ello, también se denominan fibromatosis agresivas. Los tumores pueden llegar a ser de gran tamaño y crecer agresivamente en los músculos, tendones, articulaciones e incluso huesos circundantes.
Aunque los tumores desmoides suelen crecer lentamente, en algunos casos pueden crecer rápidamente, simulando un proceso maligno o canceroso.
Los tumores se encuentran con mayor frecuencia en el hombro y la nalga, seguidos del brazo, el antebrazo y la parte posterior del muslo y la rodilla.
Los tumores desmoides pueden ocurrir a cualquier edad, pero es más probable que ocurran en niños mayores y adultos jóvenes de hasta 40 años. Los hombres se ven afectados con más frecuencia que las mujeres.
Se desconoce la causa exacta de los tumores desmoides.
Por lo general, los tumores se desarrollan en una sola área, aunque a veces pueden aparecer en varios lugares. Múltiples localizaciones de la enfermedad pueden asociarse con poliposis adenomatosa familiar, una anomalía genética en la que se desarrollan pólipos potencialmente malignos en el revestimiento intestinal.
Esto apoya la teoría de que los desmoides pueden tener una causa genética. También se han explorado otras teorías, incluidas las causas hormonales y traumáticas, pero no hay conclusiones definitivas.
Más comúnmente, un tumor desmoide causa una masa o bulto firme y levemente doloroso. A veces, la lesión puede crecer rápidamente, causando dolor y restringiendo el movimiento en las articulaciones cercanas.
Aunque rara vez son mortales, los tumores desmoides pueden invadir los nervios y los vasos sanguíneos locales. Esto puede resultar en dolor y discapacidad marcada.
Su médico realizará un examen físico completo y utilizará estudios de imágenes y otras pruebas para diagnosticar un tumor desmoide extraabdominal.
Durante el examen, su médico evaluará el tamaño y la consistencia del bulto, así como su efecto sobre la piel que lo recubre, la longitud de las extremidades y el movimiento de las articulaciones circundantes.
Imagen de resonancia magnética (IRM). Una resonancia magnética proporciona imágenes claras de los tejidos blandos y puede ser útil para evaluar los tumores desmoides. Una resonancia magnética mostrará el tamaño general y la ubicación del tumor y si alguna estructura vital circundante está involucrada.

Biopsia. Es necesaria una biopsia para confirmar el diagnóstico. Durante una biopsia, se toma una muestra de tejido del tumor y se examina bajo un microscopio. El procedimiento a menudo se realiza en el consultorio del médico. Su médico puede administrarle un anestésico local para adormecer el área y tomar una muestra con una aguja. Las biopsias también se pueden realizar como una operación pequeña.
Los tumores desmoides extraabdominales son impredecibles. Algunos tumores crecen a un ritmo rápido. Otros tumores permanecen sin cambios después del diagnóstico. Incluso hay casos de regresión espontánea. Este comportamiento errático hace que el tratamiento sea un desafío.
El tratamiento de un tumor depende de su ubicación y características, así como de las preferencias del paciente.
Se han utilizado tratamientos no quirúrgicos para tratar los tumores desmoides con resultados mixtos.
Observación. Debido a que algunos tumores desmoides crecen lentamente e incluso pueden encogerse, la primera línea de tratamiento suele ser una observación cuidadosa a lo largo del tiempo. Durante las visitas periódicas a su médico, se utilizarán estudios por imágenes y otras pruebas para detectar cambios en el tumor.
Radiación. La radioterapia puede destruir las células cancerosas y reducir los tumores. En algunos casos, los tumores desmoides se pueden tratar con éxito solo con radioterapia. En otros casos, la radioterapia se usa junto con la cirugía. Los tratamientos de radiación generalmente comienzan dos semanas después del procedimiento quirúrgico.
La radioterapia generalmente se usa solo si el paciente ha alcanzado la madurez esquelética. Existen riesgos asociados con la radiación, que incluyen fibrosis, necrosis de la piel, rigidez de las articulaciones, osteonecrosis, alteración de la placa de crecimiento y daño neurovascular. En algunos casos, la radiación en realidad puede promover el crecimiento de tumores cancerosos o sarcomas.
Los tumores desmoides tratados con una combinación de cirugía y radiación o con radioterapia sola pueden reaparecer.
Terapia hormonal. Se sabe que los desmoides tienen receptores de estrógeno. Esto significa que su crecimiento puede ser estimulado por la hormona estrógeno. Los medicamentos que bloquean la estimulación hormonal del tumor pueden ser efectivos para controlar el crecimiento del tumor.
Medicamentos antiinflamatorios no esteroideos (AINE). Los medicamentos como el ibuprofeno y el naproxeno, así como un tipo especial de AINE llamado inhibidor de la COX-II, pueden ser efectivos para tratar los tumores desmoides.
Quimioterapia. Las dosis bajas de medicamentos de quimioterapia son eficaces en el tratamiento de los desmoides. Sin embargo, el uso a largo plazo de estos medicamentos es difícil debido a sus efectos acumulativos en el cuerpo.
Otros tratamientos no quirúrgicos. La ablación por radiofrecuencia, en la que el tumor se calienta con una corriente eléctrica de alta frecuencia, así como la inyección directa del tumor con sustancias químicas irritantes (es decir, ácido acético), también se han mostrado prometedoras en el tratamiento de tumores desmoides.
En algunos casos, se puede recomendar una cirugía para extirpar el tumor. Sin embargo, puede ser difícil determinar la extensión externa del tumor durante la cirugía. Esto se debe a que los desmoides no están bien encapsulados y, a menudo, se entrelazan con otros tejidos. Un tumor puede recidivar después de la cirugía con tasas locales de recurrencia que oscilan entre el 25 % y el 68 %.
Los desmoides invaden con frecuencia los nervios y vasos sanguíneos vitales de una extremidad. Debido a esto, la extirpación completa del tumor puede afectar la función en el área circundante. La cirugía limitada, combinada con tratamientos adicionales, como radiación o quimioterapia, puede tener éxito en estos casos.
El tiempo que tardará en volver a sus actividades diarias variará según la ubicación del tumor y la extensión del procedimiento quirúrgico.
Un fisioterapeuta le proporcionará ejercicios específicos para ayudar a fortalecer sus músculos. El objetivo de la fisioterapia es ayudarlo a recuperar la función y el rango de movimiento en la extremidad afectada y las articulaciones circundantes. Los ejercicios se pueden continuar en casa o en un gimnasio local.
Continuará haciendo visitas de seguimiento a su médico cada tres meses durante dos años y luego en intervalos de 6 a 12 meses. Durante estas visitas, su médico controlará su progreso y verificará la recurrencia del tumor. Es posible que ordene pruebas como una resonancia magnética o una tomografía computarizada para verificar su progreso. Si está tomando un medicamento a largo plazo para tratar su tumor, como terapia hormonal o AINE, su médico también puede ordenar análisis de sangre.
La displasia fibrosa es rara y representa aproximadamente el 7 % de todos los tumores óseos benignos. Puede afectar cualquier hueso del cuerpo, pero ocurre con mayor frecuencia en:
La displasia fibrosa se ha relacionado con una mutación genética que hace que las células óseas produzcan un tipo anormal de hueso fibroso. Aunque el hueso anormal comienza a formarse antes del nacimiento, su presencia a menudo no se descubre hasta la niñez, la adolescencia o incluso la edad adulta.
La condición generalmente se divide en dos tipos:
La displasia fibrosa es un trastorno crónico y, a menudo, es progresivo. Aunque las lesiones pueden estabilizarse y dejar de crecer, no desaparecen. Las lesiones individuales pueden progresar más rápidamente en la forma poliostótica de la afección y en los niños en crecimiento.
En pacientes con displasia fibrosa, la misma anomalía que ocurre en las células óseas también puede ocurrir en las células de las glándulas del cuerpo, lo que lleva a anomalías hormonales. Aunque esto es raro, puede ocurrir en formas graves de displasia fibrosa poliostótica.
La displasia fibrosa puede ocurrir como parte de un trastorno más grande. Por ejemplo, el síndrome de McCune-Albright se caracteriza por displasia fibrosa poliostótica que se presenta con anormalidades hormonales y áreas de piel oscurecida (manchas “café con leche”).
Es muy raro que las áreas de displasia fibrosa se vuelvan malignas o cancerosas. Esto ocurre en menos del 1% de los pacientes y es más probable que suceda en pacientes con la forma poliostótica de la afección o en pacientes con el síndrome de McCune-Albright.
Se desconoce la causa de la mutación del gen. No se hereda y no se sabe si es causada por la dieta o factores ambientales. La mutación ocurre por igual en hombres y mujeres de todas las razas.
En algunos casos, las lesiones son indoloras y no causan síntomas. Cuando esto sucede, la condición se puede encontrar inesperadamente cuando se realizan radiografías o pruebas para una lesión o condición médica no relacionada. Cuando se presentan síntomas, reflejan el tamaño y la gravedad de la lesión.
A medida que el tejido óseo fibroso crece y se expande, el área afectada puede volverse débil y dolorosa. Es más probable que se produzca dolor si el hueso afectado es uno de los huesos de la pierna o la pelvis que soportan peso.
El dolor causado por la displasia fibrosa generalmente comienza como un dolor sordo que empeora con la actividad y disminuye con el descanso. Puede empeorar progresivamente con el tiempo.
El hueso fibroso es muy débil en comparación con el hueso normal. A veces puede romperse o fracturarse a través del área debilitada y causar un dolor repentino e intenso. Una fractura a menudo ocurre después de un período de dolor sordo, aunque también puede ocurrir repentinamente sin ningún dolor previo.

En pacientes que experimentan fracturas repetidas, la cicatrización deficiente puede provocar una deformidad ósea. Si esta deformidad ocurre en los huesos faciales o resulta en una curvatura de los huesos de las piernas, puede volverse muy notoria.
La deformidad severa en los huesos faciales puede conducir a la pérdida de la visión o la audición. Si las piernas o la pelvis están involucradas, el paciente puede tener problemas para caminar o desarrollar artritis en las articulaciones cercanas.


Los pacientes con anomalías hormonales pueden experimentar una serie de síntomas.
Los pacientes jóvenes con anomalías hormonales pueden desarrollar una pubertad temprana. Este problema es más común en niñas que en niños y es causado por la hiperactividad de los ovarios. La hiperactividad también puede ocurrir en otras glándulas del cuerpo, incluyendo:
Las mujeres pueden experimentar un aumento del dolor durante el embarazo o el ciclo menstrual, ya que los niveles elevados de hormonas durante estos períodos pueden acelerar el crecimiento de la displasia fibrosa.
Las manchas cutáneas pigmentadas a menudo se observan en pacientes con anomalías hormonales y displasia fibrosa.

En muy raras ocasiones, las áreas de displasia fibrosa pueden volverse cancerosas. Cuando esto ocurre, las señales de advertencia pueden incluir un rápido aumento de la inflamación o el crecimiento de una lesión. Los pacientes también pueden experimentar un dolor cada vez mayor, especialmente el dolor que los despierta por la noche o que no desaparece con el descanso.
Durante el examen, su médico hablará con usted sobre su salud general y su historial médico y le preguntará acerca de sus síntomas.
Las radiografías y otras pruebas ayudarán a su médico a confirmar el diagnóstico de displasia fibrosa o determinar la extensión del trastorno.
Rayos X. Los rayos X proporcionan imágenes de estructuras densas como los huesos. En un paciente con displasia fibrosa, una radiografía puede mostrar:

Aunque una fractura generalmente se puede ver en una radiografía y/o resonancia magnética, una tomografía computarizada a veces puede ayudar a su médico a ver mejor la fractura y determinar la calidad del hueso.
Radiografía. Su médico puede ordenar una gammagrafía ósea para buscar lesiones adicionales en todo su esqueleto. Durante esta prueba, se inyecta una cantidad muy pequeña de colorante radiactivo en el cuerpo por vía intravenosa. La exploración mostrará un “punto caliente”, un aumento de la absorción del material radiactivo, en áreas de hueso anormal.
Pruebas de laboratorio. Cuando las lesiones de displasia fibrosa están creciendo activamente, los análisis de sangre pueden mostrar niveles elevados de la enzima fosfatasa alcalina. Los análisis de orina tienden a mostrar niveles elevados de hidroxiprolina. Sin embargo, estas anomalías no son específicas de la displasia fibrosa y no siempre están presentes. A veces se pueden ver en otras condiciones médicas que involucran el crecimiento óseo, e incluso se pueden ver en el crecimiento óseo normal.
Biopsia. Puede ser necesaria una biopsia para confirmar el diagnóstico de displasia fibrosa. En una biopsia, se toma una muestra de tejido de la lesión y se examina bajo un microscopio.
Una biopsia se puede realizar bajo anestesia local con una aguja o como una pequeña operación abierta.
Observación. Si sus lesiones no causan síntomas, su médico puede recomendar observarlas y monitorearlas para ver si progresan. Durante este tiempo, es posible que necesite radiografías periódicas u otras pruebas.
Medicamentos
Los bisfosfonatos son medicamentos que disminuyen la actividad de las células que disuelven el hueso. Recientemente han estado disponibles en forma de píldoras fáciles de tomar. Estos medicamentos aún no se han usado ampliamente en el tratamiento de la displasia fibrosa; sin embargo, los primeros estudios han mostrado un alivio efectivo del dolor asociado con el trastorno.
Vigorizante. En algunos casos, se pueden usar aparatos ortopédicos para prevenir fracturas en huesos debilitados. Sin embargo, no se ha demostrado que los aparatos ortopédicos sean efectivos para prevenir la progresión de la deformidad.
En pacientes con displasia fibrosa, a menudo es necesaria la cirugía para extirpar un crecimiento o reparar o prevenir fracturas óseas. Su médico puede recomendarle una cirugía si tiene:
Legrado. El legrado es un procedimiento quirúrgico comúnmente utilizado para tratar la displasia fibrosa. En el legrado, se raspa el tumor del hueso.
Injerto óseo. Después del legrado, el médico puede llenar la cavidad con un injerto óseo para ayudar a estabilizar el hueso. Un injerto óseo es hueso extraído de un donante (aloinjerto) o de otro hueso del cuerpo (autoinjerto), generalmente de la cadera. También se puede utilizar material óseo sintético para rellenar la cavidad.
En algunos casos, el injerto óseo colocado en la cavidad puede reabsorberse y la displasia fibrosa puede reaparecer.
Fijación interna. Se pueden usar barras o placas de metal y tornillos para reparar una fractura o deformidad, prevenir la rotura del hueso antes de que ocurra o estabilizar el hueso.

La investigación genética en curso puede conducir a una mejor comprensión de la mutación genética exacta involucrada en la displasia fibrosa. Entonces, los médicos podrán desarrollar tratamientos no quirúrgicos más efectivos.
Las nuevas generaciones de medicamentos, incluidos los bisfosfonatos, ahora son más fáciles de tomar y tienen menos efectos secundarios. Más experiencia con estos medicamentos también puede permitir un tratamiento más eficaz de la displasia fibrosa.
Los tumores de células gigantes reciben su nombre por la forma característica en que se ven cuando se observan bajo el microscopio. Cuando se observan al microscopio, los tumores consisten en muchas células inusualmente grandes o “gigantes”. Estas células están formadas por la fusión de varias células individuales en una sola célula más grande.

Muchos tipos de tumores óseos y otras afecciones (incluido el hueso normal) contienen células gigantes. El diagnóstico de tumor óseo de células gigantes se realiza cuando se observa una gran cantidad de células gigantes entre un fondo de otras células anormales.
Si bien la mayoría de los tumores óseos ocurren en el área ensanchada cerca de los extremos de los huesos largos del cuerpo (metáfisis), los tumores de células gigantes ocurren casi exclusivamente en la porción final de los huesos largos (epífisis), directamente al lado de las articulaciones.

Con mayor frecuencia, los tumores ocurren cerca de la articulación de la rodilla, ya sea en el extremo inferior del fémur (fémur) o en el extremo superior de la espinilla (tibia).
Otras ubicaciones comunes incluyen:
En casos raros, un paciente puede tener múltiples tumores de células gigantes en diferentes huesos, una condición llamada “tumor óseo de células gigantes multicéntrico”.
La mayoría de los tumores de células gigantes ocurren en pacientes entre 20 y 40 años de edad. Por lo general, no ocurren en niños o en adultos mayores de 65 años. Ocurren un poco más a menudo en las mujeres.
Si bien los tumores de células gigantes suelen ser benignos (no cancerosos), pueden crecer rápidamente y destruir el hueso cercano a una articulación. En casos raros, un tumor de células gigantes puede diseminarse o hacer metástasis a los pulmones.
Se desconoce la causa de los tumores de células gigantes. Los tumores ocurren espontáneamente. No se sabe que sean causados por traumatismos, factores ambientales o dieta. Los tumores óseos de células gigantes no se heredan.
Los tumores también pueden estar asociados con hiperactividad de las glándulas paratiroides, una condición conocida como “hiperparatiroidismo”. Esto es diferente a un tumor óseo aislado de células gigantes.
El síntoma más común de un tumor de células gigantes es el dolor en el área del tumor. El paciente también puede tener dolor con el movimiento de la articulación cercana. Este dolor generalmente aumenta con la actividad y disminuye con el reposo.
El dolor suele ser leve al principio, pero empeora con el tiempo a medida que el tumor aumenta de tamaño. En ocasiones, el hueso debilitado por el tumor se rompe y provoca la aparición repentina de un dolor intenso.
A veces, el paciente no tendrá dolor en absoluto, pero notará una masa o un área hinchada en su lugar.
Su médico realizará un examen físico completo y utilizará radiografías y otras pruebas para diagnosticar un tumor de células gigantes.
Radiografía. Este estudio proporciona imágenes de estructuras densas, como el hueso. Su médico ordenará una radiografía para ayudar a confirmar el diagnóstico.
En la radiografía, un tumor de células gigantes aparece como una lesión destructiva (lítica) junto a una articulación. Ocasionalmente, el área afectada del hueso estará rodeada por un borde delgado de hueso blanco, que puede ser completo o incompleto. También puede haber expansión del área afectada del hueso.


Otros estudios de imagen. Su médico puede ordenar una resonancia magnética nuclear (RMN) o una tomografía computarizada (TC) para evaluar mejor el tumor y el área que lo rodea.
También se puede realizar una tomografía computarizada o una radiografía de tórax en el momento del diagnóstico inicial para determinar si el tumor se ha diseminado a los pulmones.

Radiografía. En algunos casos, su médico puede ordenar una gammagrafía ósea. Durante esta prueba, se inyecta una cantidad muy pequeña de colorante radiactivo en el cuerpo por vía intravenosa. Los tumores de células gigantes generalmente provocan una mayor captación del material radiactivo y aparecen como un “punto caliente” en el hueso donde se encuentra el tumor.
Biopsia. Es posible que se necesite una biopsia para confirmar el diagnóstico de un tumor de células gigantes. En una biopsia, se toma una muestra de tejido del tumor y se examina bajo un microscopio.
Una biopsia se puede realizar bajo anestesia local con una aguja o como una pequeña operación abierta.
Sin tratamiento, un tumor de células gigantes continuará creciendo y destruyendo el hueso circundante, por lo que siempre es necesario el tratamiento. Los objetivos del tratamiento son:
La cirugía es el tratamiento preferido para los tumores de células gigantes; sin embargo, hay algunos casos en los que un tumor no se puede extirpar de manera segura o eficaz mediante cirugía. En este caso, su médico puede recomendar un tratamiento no quirúrgico.
El tratamiento no quirúrgico puede incluir:
Radiación. A veces, la radioterapia se puede usar para reducir los tumores de células gigantes en áreas donde la cirugía puede ser difícil de realizar sin dañar los tejidos sensibles, como la columna vertebral. Sin embargo, la radioterapia puede provocar la formación de cáncer en algunos pacientes, por lo que se usa solo en casos excepcionales.
Embolización tumoral. Durante este procedimiento, se bloquean arterias específicas que suministran sangre al tumor. Sin su suministro de oxígeno y nutrientes, las células tumorales comienzan a morir. La mayoría de las veces, la embolización se realiza antes de la cirugía, pero también se puede usar sola en los casos en que no se puede realizar la cirugía.
Medicamento. La FDA aprobó recientemente el uso de un medicamento inyectable para el tratamiento de tumores de células gigantes. El medicamento funciona dirigiéndose a un receptor especial en las células gigantes. Esto disminuye la actividad y ralentiza la descomposición del hueso. Sin embargo, debido a que las células tumorales están ubicadas entre las células gigantes, el tumor reaparecerá después de suspender el tratamiento. El medicamento a veces se usa en casos en los que no se puede realizar una cirugía o para tumores recurrentes.
La cirugía ha demostrado ser el tratamiento más efectivo para los tumores de células gigantes. El tratamiento quirúrgico puede incluir:
Legrado. El curetaje es el procedimiento quirúrgico más utilizado para tratar los tumores de células gigantes. En el legrado, se utilizan instrumentos especiales para raspar el tumor del hueso.
Injerto óseo. Después del curetaje, la cavidad se llena con un injerto óseo para ayudar a estabilizar el hueso. Un injerto óseo es hueso que se toma de un donante (aloinjerto) o de otro hueso de su propio cuerpo (autoinjerto), generalmente de la cadera.
Su médico también puede usar una mezcla de cemento óseo para rellenar el orificio. Por lo general, se colocan productos químicos adicionales, como nitrógeno líquido, peróxido de hidrógeno o fenol, dentro de la cavidad ósea para tratar de reducir el riesgo de recurrencia. En algunos casos, se utilizará un láser de gas argón para ayudar a reducir el riesgo de recurrencia.

Otros procedimientos. Si un tumor ha reaparecido o ha causado un daño óseo o tisular excesivo, es posible que sea necesaria una extirpación quirúrgica y una reconstrucción más complejas.
Su médico puede usar injertos óseos, articulaciones artificiales o una combinación de estos para reconstruir los sitios de hueso, articulación o tejido blando. El objetivo es restaurar la parte del cuerpo para que el paciente pueda realizar sus actividades cotidianas normales.
En casos raros, un tumor de células gigantes puede diseminarse a los pulmones. Si el tumor se propaga, es necesaria la extirpación quirúrgica del hueso, así como del área afectada del pulmón. Esto típicamente resultará en una cura.
Su resultado después del tratamiento dependerá de una serie de factores, que incluyen:
Los tumores de células gigantes pueden reaparecer, por lo que es importante que consulte a su médico para visitas de seguimiento periódicas durante varios años después del tratamiento. Durante estas visitas, su médico tomará radiografías del sitio afectado, así como radiografías de tórax.
Un hemangioma ocurre cuando los vasos sanguíneos pequeños comienzan a multiplicarse a un ritmo anormal y forman una masa o un bulto. Es posible tener más de un hemangioma.
Hay varios tipos de hemangioma. Algunos de los tipos más comunes se describen a continuación:
Los hemangiomas de la piel son comunes en los bebés. A veces están presentes al nacer, pero lo más habitual es que aparezcan en las primeras semanas o meses de vida. La mayoría de los hemangiomas infantiles son hemangiomas capilares, aunque también existen tipos cavernosos y compuestos. Las niñas se ven afectadas un poco más a menudo que los niños.
Los hemangiomas infantiles comunes siguen el mismo patrón de crecimiento: un período de crecimiento rápido, a menudo durante el primer año, seguido de un período de reducción del tamaño del tumor (llamado involución o regresión). El tiempo que tarda el tumor en alcanzar su tamaño completo y luego encogerse varía mucho, pero la mayoría de los hemangiomas infantiles han terminado de involucionar cuando el niño llega a la pubertad.
La mayoría de estos hemangiomas se reducirán completamente por sí solos y no requerirán tratamiento. Sin embargo, algunos hemangiomas pueden causar problemas con funciones vitales como respirar, comer y ver, y requieren algún tipo de tratamiento. Además, debido a que estos tumores pueden llegar a ser grandes ya menudo aparecen en la cara, el cuello y el cuero cabelludo, se deben considerar las necesidades emocionales del niño al determinar las opciones de tratamiento.
Los hemangiomas que están presentes al nacer (llamados hemangiomas congénitos) siguen un patrón de crecimiento diferente. Estos hemangiomas están completamente desarrollados al nacer e involucionan por completo durante el primer año del bebé (llamado hemangioma congénito de involución rápida) o no involucionan en absoluto (llamado hemangioma congénito no involutivo).
Aunque no son tan comunes como los hemangiomas de la piel, los hemangiomas se desarrollan en otros tejidos, incluidos los músculos y los huesos.
Hemangioma intramuscular. Los hemangiomas en el tejido muscular pueden desarrollarse a cualquier edad, pero ocurren con mayor frecuencia en adultos jóvenes. Los hemangiomas capilares son más comunes en el músculo que los tipos cavernosos y compuestos. Cualquier músculo puede estar involucrado.
Debido a que están ubicados dentro del músculo, estos hemangiomas a menudo no muestran signos visibles, aunque algunos pueden causar inflamación en el área del tumor que aumenta con la actividad. Estos tumores a menudo son dolorosos y requieren tratamiento.
Hemangioma óseo. Los hemangiomas que ocurren en los huesos generalmente ocurren en el cráneo o la columna vertebral y son más comunes en personas de 50 a 70 años de edad. Los tipos capilar y cavernoso son los hemangiomas más comunes que se encuentran en los huesos. Pueden crecer en la superficie o más profundamente en el canal central de un hueso. Debido a que por lo general no causan síntomas, estos tumores a menudo se encuentran por casualidad cuando se toma una imagen de rayos X o MRI para otro propósito.
Hemangioma de órgano interno . Aunque es poco común, los hemangiomas pueden desarrollarse en los órganos internos, con mayor frecuencia en el hígado y los intestinos. Al igual que los hemangiomas que se encuentran en los huesos, los hemangiomas en los órganos internos a menudo se encuentran por casualidad durante las pruebas con otro propósito. A menos que estén causando síntomas, estos hemangiomas no requieren ningún tratamiento.
Los hemangiomas a menudo se confunden con malformaciones vasculares, que son grupos de vasos sanguíneos que se desarrollan en arterias, venas, capilares o vasos linfáticos. Los tipos comunes de malformaciones vasculares incluyen malformaciones arteriovenosas (MAV) y ectasias vasculares.
Las malformaciones vasculares ocurren durante el desarrollo fetal. Aunque están presentes al nacer, algunos tipos de malformaciones vasculares pueden no causar síntomas hasta la edad adulta.
Las malformaciones vasculares difieren de los hemangiomas infantiles en que crecen lentamente junto con el niño y se vuelven más evidentes con la edad. Debido a que no se reducen ni se resuelven por sí solos, muchos tipos de malformaciones vasculares en los niños requieren tratamiento.
Los diferentes tipos de hemangiomas se asocian con diferentes causas, aunque no se comprende bien por qué exactamente se desarrollan los hemangiomas. Por ejemplo, los hemangiomas infantiles son causados por errores en el desarrollo del sistema vascular que ocurren durante el desarrollo fetal pero, en muchos casos, no se puede identificar el evento que causó el error. Algunos hemangiomas se desarrollan después de una lesión, pero no se ha probado si una lesión realmente puede causar un hemangioma. Algunos hemangiomas se desarrollan con el embarazo y desaparecen después. Algunos hemangiomas están asociados con anomalías genéticas (por ejemplo, hemangiomas cavernosos en la enfermedad de von Hippel-Lindau).
No ha habido conexión comprobada entre el desarrollo de hemangiomas y cualquier ocupación particular o exposición a químicos o radiación. Además, no se sabe que ningún alimento, medicamento o actividad conocida durante el embarazo cause un hemangioma en un bebé.
Los hemangiomas suelen ser lesiones indoloras, de color rojo a azul, en la piel, los labios o dentro de la boca. Suelen ser suaves al tacto. La mayoría de las veces están al ras de la piel o ligeramente elevados, pero a veces crecen a partir de un tallo. Las lesiones superficiales pueden sangrar o convertirse en llagas, especialmente si se golpean o lesionan.
Los hemangiomas profundos en el músculo pueden causar dolor, así como inflamación alrededor del hemangioma que aumenta con la actividad.
Los hemangiomas en los huesos pueden causar dolor y agrandamiento del hueso.
Los hemangiomas se pueden confundir con otras malformaciones vasculares. Debido a que el tratamiento puede variar considerablemente dependiendo de si la lesión es un hemangioma u otro tipo de malformación, se recomienda un examen médico completo.
Antes de un examen físico, su médico hablará con usted sobre su salud general y su estado actual, o el de su hijo, si es un padre o familiar. Él o ella querrá obtener un buen historial de la lesión, particularmente cuánto tiempo ha estado allí la masa y si ha cambiado con el tiempo. Su médico querrá saber qué síntomas, como el dolor, están asociados con la lesión y cuándo comenzaron los síntomas.
Durante el examen físico, su médico inspeccionará y palpará cualquier masa, observando exactamente dónde se encuentra, cómo se siente y tal vez cualquier cambio en la piel circundante.
Aunque los médicos a menudo pueden diagnosticar los hemangiomas basándose únicamente en el historial médico y el examen físico, las pruebas de imagen pueden ser útiles.
Radiografías y tomografías computarizadas (TC). Aunque estas pruebas crean mejores imágenes de estructuras densas como el hueso, las radiografías simples y las tomografías computarizadas pueden mostrar un hemangioma cavernoso si tiene calcificaciones. Estas calcificaciones se denominan flebolitos.
Imágenes por resonancia magnética (IRM). Estos escaneos pueden crear imágenes claras de estructuras blandas como un hemangioma. Los hemangiomas en una resonancia magnética a menudo se describen como una “bolsa de gusanos” debido a la forma en que los vasos sanguíneos se juntan entre sí.
angiograma. En esta prueba, se inyecta un tinte en el torrente sanguíneo circundante para que el hemangioma aparezca en una imagen de rayos X.
Biopsia. A veces, puede ser difícil distinguir los hemangiomas de otros tumores y, a veces, es necesaria una biopsia para confirmar el diagnóstico de hemangioma. En una biopsia, se toma una muestra de tejido del tumor y se examina bajo un microscopio. Mirar el tejido bajo un microscopio puede ser la mejor manera de saber si el tumor es realmente un hemangioma y de qué subtipo es.
Durante la biopsia, su médico puede administrarle un anestésico local para adormecer el área y tomar una muestra con una aguja. Las biopsias también se pueden realizar como una operación pequeña.
Análisis de sangre. Si hay múltiples tumores o si sus síntomas son preocupantes para un patrón de enfermedad específico, su médico puede recomendar análisis de sangre para un análisis genético.
Las imágenes y las pruebas de tejidos ayudan a su médico a diferenciar el hemangioma de otros tipos de malformaciones vasculares y tumores de tejidos blandos.
Al planificar el tratamiento, es muy importante diferenciar el hemangioma de los tumores vasculares cancerosos más agresivos, como el angiosarcoma.
El tratamiento varía ligeramente según el subtipo de hemangioma.
Observación. Aunque es posible que un hemangioma no requiera ningún tratamiento, es importante visitar a su médico regularmente para monitorear cualquier cambio en el tumor.
Medicamentos betabloqueantes. Los bloqueadores beta son un medicamento que se puede recomendar según el tipo y el tamaño de un hemangioma. Para ciertos tipos, como los hemangiomas infantiles y superficiales, los bloqueadores beta se pueden administrar por vía oral (en forma de píldora) con el objetivo de retardar el crecimiento de la lesión.
Medicación antiinflamatoria. Si un hemangioma está creciendo cerca de estructuras vitales, como la nariz, los labios o los párpados, su médico puede recomendarle medicamentos con esteroides. Los esteroides a menudo se usan para retrasar el crecimiento del tumor. Los medicamentos pueden inyectarse directamente en el hemangioma o administrarse por vía oral.
Compresión. La compresión neumática intermitente es un tratamiento que utiliza mangas o mallas inflables para aplicar presión al tumor. Puede ayudar a disminuir la hinchazón asociada con un hemangioma. Sin embargo, no hará que el hemangioma desaparezca.
Embolización. En este procedimiento, se cierra el suministro de sangre al tumor. Este es un procedimiento mínimamente invasivo en el que se inyectan pequeñas partículas en los vasos sanguíneos para bloquearlos. La escleroterapia es un procedimiento similar en el que se utilizan agentes químicos para cerrar los vasos. Estos procedimientos pueden ser muy útiles para reducir el tamaño del tumor y disminuir el dolor. Sin embargo, a menudo, el tumor volverá a aumentar su suministro de sangre con el tiempo después de estos procedimientos. A veces, la embolización también se usa antes de la cirugía para reducir el riesgo de una gran pérdida de sangre.
Tratamiento láser. Para algunos pacientes, los láseres pueden ser útiles para extirpar un tumor, preparar un tumor para otros tratamientos o disminuir el dolor y otros síntomas no deseados. Según el tamaño del tumor y su ubicación, es posible que se necesiten múltiples tratamientos con láser. El tratamiento con láser generalmente se reserva para los hemangiomas que afectan la piel.
Se puede recomendar la cirugía para un hemangioma cavernoso si la lesión está destruyendo los tejidos sanos que lo rodean. En algunos casos, un hemangioma puede causar síntomas dolorosos lo suficientemente graves como para considerar el tratamiento quirúrgico.
Procedimiento. El procedimiento para extirpar quirúrgicamente un hemangioma se llama escisión. Se usa anestesia general para ponerlo a dormir, luego su médico hará una incisión en su piel y extirpará el tumor.
Recuperación. Es probable que tenga puntos de sutura, que su médico retirará en unas pocas semanas. Su médico envolverá el área con un vendaje compresivo y le dará instrucciones específicas sobre las restricciones de actividad para guiar su recuperación.
Complicaciones. La complicación más común de la cirugía para extirpar un hemangioma es la hemorragia (pérdida de sangre). Además, los hemangiomas tienen una alta tendencia a reaparecer después de la cirugía, según el tipo y la ubicación del tumor.
Actualmente hay una gran cantidad de investigaciones que intentan aprender más sobre la proliferación vascular no regulada, incluidos los hemangiomas. El desarrollo de medicamentos dirigidos a detener el desarrollo de los vasos sanguíneos (antiangiogénicos) es un área de investigación apasionante para muchos tipos de tumores.
Una articulación es donde se unen los extremos de dos o más huesos, como la articulación de la rodilla, la articulación del hombro o la articulación del tobillo.
Las articulaciones sanas están cubiertas de cartílago articular, una sustancia resbaladiza que ayuda a que los huesos se deslicen suavemente unos sobre otros durante el movimiento.
Gruesas bandas de tejido rodean la articulación, formando una cápsula que la mantiene unida. La superficie inferior de la cápsula articular está revestida por una membrana delgada llamada membrana sinovial . El sinovio produce un líquido que lubrica la articulación.

En la condromatosis sinovial, la sinovial crece de manera anormal y produce nódulos hechos de cartílago. Estos nódulos a veces pueden desprenderse de la membrana sinovial y aflojarse dentro de la articulación.
El tamaño de los cuerpos cartilaginosos sueltos dentro de la articulación puede variar, desde unos pocos milímetros (el tamaño de una pastilla pequeña) hasta unos pocos centímetros (el tamaño de una canica).
El líquido sinovial nutre los cuerpos sueltos y pueden crecer, calcificarse (endurecerse) u osificarse (convertirse en hueso). Cuando esto ocurre, pueden rodar libremente dentro del espacio articular.
A medida que ruedan, los cuerpos sueltos pueden dañar el cartílago articular liso que cubre la articulación y causar osteoartritis. En la osteoartritis, el cartílago dañado se desgasta y se deshilacha. Mover los huesos a lo largo de esta superficie articular expuesta es doloroso.
En casos severos de condromatosis sinovial, los cuerpos sueltos pueden crecer lo suficiente como para ocupar todo el espacio articular o penetrar en los tejidos adyacentes.

La condromatosis sinovial ocurre con mayor frecuencia en la rodilla, seguida de la cadera, el codo y el hombro. En la mayoría de los casos, solo se ve afectada una articulación del cuerpo.
La afección ocurre con mayor frecuencia en pacientes de entre 30 y 50 años. Los hombres se ven afectados con el doble de frecuencia que las mujeres.
Se desconoce la causa de la condromatosis sinovial. La condición ocurre espontáneamente. No se hereda.
Los síntomas más comunes de la condromatosis sinovial son similares a los de la osteoartritis. Éstas incluyen:
Otros signos y síntomas pueden incluir:
Los nódulos a veces se pueden sentir en las articulaciones que están cerca de la piel, como las articulaciones de la rodilla, el tobillo y el codo.
Es importante buscar tratamiento para la condromatosis sinovial lo antes posible para ayudar a aliviar los síntomas dolorosos y prevenir la progresión de la osteoartritis en la articulación.
Su médico hablará con usted sobre su salud general y su historial médico y le preguntará acerca de sus síntomas. Él o ella luego examinará cuidadosamente la articulación afectada, en busca de:
Su médico ordenará estudios de imágenes para ayudar a diagnosticar la condromatosis sinovial. Los estudios de imágenes también ayudarán a su médico a diferenciar la condromatosis sinovial de la osteoartritis.
Rayos X. Los rayos X proporcionan imágenes de estructuras densas, como los huesos. Los cuerpos sueltos más grandes suelen estar calcificados u osificados y pueden verse en las radiografías. Los cuerpos sueltos más pequeños y los que no están calcificados u osificados pueden no aparecer.
Otros estudios de imagen. Si los cuerpos sueltos no son visibles en la radiografía, su médico puede solicitar una resonancia magnética nuclear (RMN) o una tomografía computarizada (TC) para evaluar mejor la articulación. Los cuerpos sueltos generalmente se pueden ver tanto en la resonancia magnética como en la tomografía computarizada.


Además de mostrar los cuerpos sueltos, las pruebas de imágenes también pueden ayudar a su médico a determinar si tiene algún problema adicional, como líquido en la articulación o signos de osteoartritis (estrechamiento del espacio articular y espolones óseos).
Observación. Dependiendo de sus síntomas, la simple observación a veces puede ser una opción de tratamiento. Su médico considerará cuidadosamente una serie de factores para determinar si la observación es apropiada en su caso.
Durante este tiempo, su médico controlará de cerca la articulación afectada para verificar la progresión de la osteoartritis.
El tratamiento para la condromatosis sinovial generalmente implica cirugía para extirpar los cuerpos sueltos de cartílago. En algunos casos, la membrana sinovial también se extirpa parcial o totalmente (sinovectomía) durante la cirugía.
La cirugía se puede realizar mediante un procedimiento abierto o un procedimiento artroscópico. La técnica que use su médico dependerá de una serie de factores, que incluyen:

En un procedimiento abierto tradicional, el médico generalmente hará una o dos incisiones grandes.
En un procedimiento artroscópico, el médico hará incisiones más pequeñas y utilizará instrumentos quirúrgicos en miniatura para extraer los cuerpos sueltos.
Los resultados finales de los procedimientos abiertos y artroscópicos son los mismos. Su médico hablará con usted sobre qué técnica quirúrgica es mejor en su caso.

El tiempo que tarde en volver a sus actividades diarias variará, según el tipo de procedimiento que tenga. Su médico le proporcionará instrucciones específicas para guiar su rehabilitación.
La condromatosis sinovial puede regresar hasta en un 20 por ciento de los pacientes. Durante un período de tiempo después de la cirugía, su médico programará visitas de seguimiento periódicas para detectar cualquier recurrencia.
Su médico también controlará la articulación para detectar cualquier progresión de la osteoartritis. La cantidad de daño que la condromatosis sinovial ya le haya causado a la articulación influirá en su probabilidad de desarrollar osteoartritis.
Dentro de la médula ósea normal, los glóbulos rojos, los glóbulos blancos y las plaquetas inmaduros se multiplican, maduran y se liberan al torrente sanguíneo. En algunos casos, sin embargo, las mutaciones genéticas provocan cambios en estas células que pueden alterar este proceso.
En los pacientes con leucemia, algunos de los glóbulos blancos de la médula ósea no maduran por completo, por lo que no funcionan correctamente. Estas células anormales crecen más rápidamente que las células normales y continúan creciendo cuando deberían detenerse. Con el tiempo, comienzan a saturar la médula ósea, impidiendo el funcionamiento normal y el crecimiento de células sanguíneas sanas.
La leucemia se describe como aguda o crónica, según la rapidez con la que progresa:
La leucemia se clasifica además según el tipo de glóbulo blanco que afecta:
Hay cuatro tipos principales de leucemia:
La leucemia linfoblástica aguda es el tipo más común y representa alrededor del 80 por ciento de todos los casos. Ocurre con mayor frecuencia en niños y adolescentes.
Los otros tres tipos (LLL, AML y CML) ocurren con mayor frecuencia en adultos, generalmente en pacientes que tienen entre 50 y 60 años.
Se desconoce la causa de la leucemia. Sin embargo, algunas cosas pueden aumentar su riesgo de desarrollarlo, como tener ciertas condiciones hereditarias, estar expuesto a ciertas toxinas o tener ciertos tipos de infecciones virales.
Los síntomas de la leucemia varían según el tipo específico; sin embargo, muchos pacientes experimentan:
Otros signos y síntomas pueden incluir:
Aunque es raro, algunos pacientes pueden desarrollar fracturas óseas patológicas si un tumor debilita el hueso. Esto es más común si el tumor se desarrolla en un área del esqueleto que soporta peso, como la pelvis, la columna vertebral o el fémur (hueso del muslo).
Su médico hablará con usted sobre su salud general y su historial médico y le preguntará acerca de sus síntomas. Él o ella luego realizará un examen físico, en busca de:
Su médico usará análisis de sangre para ayudar a diagnosticar la leucemia.
Se utilizarán varias pruebas diferentes para analizar su muestra de sangre. Estas pruebas incluyen:
Otras anomalías que pueden identificarse con análisis de sangre incluyen anemia (recuento bajo de glóbulos rojos) o trombocitopenia (recuento bajo de plaquetas).
Identificar la etapa o la gravedad de su leucemia ayudará a su médico a determinar la estrategia de tratamiento más eficaz.
La estadificación está determinada por una serie de factores, que incluyen:
El tratamiento de la leucemia por lo general se lleva a cabo en tres fases. Cada fase involucra diferentes combinaciones de quimioterapia y medicamentos. Su médico considerará varios factores al determinar las terapias apropiadas en cada fase, que incluyen:
Las tres fases del tratamiento de la leucemia generalmente incluyen:
Inducción de la remisión. Durante la primera fase del tratamiento, se usan varios medicamentos contra el cáncer para combatir y destruir la mayor cantidad posible de células anormales, con el objetivo de hacer que el cáncer entre en remisión. Esta fase puede llevar meses, pero por lo general termina en un año.
Terapia de consolidación. Una vez que se logra la remisión, el tratamiento se enfoca en prevenir la propagación del cáncer a otras partes del cuerpo. Las terapias utilizadas en esta fase pueden incluir rondas adicionales de quimioterapia, otros medicamentos específicos para la leucemia o trasplante de médula ósea con células madre. Esta fase suele completarse en menos de dos años.
Terapia de mantenimiento. La tercera fase del tratamiento se enfoca en mantener la remisión del cáncer a través de una combinación de medicamentos y ciclos de quimioterapia. La duración de la terapia de mantenimiento varía según la gravedad de la leucemia.
Los tratamientos novedosos a través de estudios clínicos también pueden estar disponibles para pacientes que cumplan con ciertos criterios de selección. Su médico hablará con usted sobre el mejor curso de tratamiento para su situación específica.
Su resultado después del tratamiento dependerá de una serie de factores, que incluyen:
Después del tratamiento, se reunirá con su médico para que pueda evaluar su respuesta a la terapia. También se programará citas regulares de seguimiento para que su médico pueda buscar signos de complicaciones del tratamiento o recurrencia. La recurrencia de la leucemia es posible incluso muchos años después de haber terminado el tratamiento. Por esta razón, la mayoría de los pacientes necesitarán muchos años de seguimiento.
El pronóstico de los pacientes con leucemia es muy variable. Incluso dentro de tipos específicos, algunos pacientes responden bien a ciertos tratamientos mientras que otros no. Su médico hablará con usted sobre su pronóstico específico.
Si bien todos los lipomas se componen de grasa, existen subtipos basados en la forma en que aparecen bajo el microscopio. Algunas variedades incluyen:
La causa de los lipomas no se entiende completamente. Algunos subtipos parecen tener un defecto genético (lipomas convencionales, lipomas de células fusiformes, lipomas pleomórficos) y se pueden heredar de miembros de la familia.
No ha habido conexión comprobada entre el desarrollo de lipomas y cualquier ocupación particular o exposición a químicos o radiación. Algunos médicos creen que los lipomas ocurren con mayor frecuencia en personas inactivas.
Los lipomas suelen ser masas redondeadas que se sienten suaves y gomosas. Los lipomas justo debajo de la piel se pueden mover con un empujón suave. Los lipomas no suelen doler, aunque algunos de los subtipos pueden ser dolorosos, como el angiolipoma.
A menudo lleva más tiempo notar los lipomas que están en el tejido más profundo y estos tumores pueden ser bastante grandes cuando realmente se descubren. Los lipomas más profundos también tienden a ser menos móviles.
Antes de un examen físico, su médico hablará con usted sobre su estado de salud general, así como sobre su estado actual. Él o ella querrá obtener un buen historial del problema desde su perspectiva, particularmente cuánto tiempo ha estado allí la masa y qué síntomas, como el dolor, están asociados con ella.
Durante el examen físico, su médico palpará la masa, comprobando su tamaño y consistencia, así como su movilidad. Él o ella también examinará la piel que recubre la masa, en busca de cambios.
Aunque los médicos generalmente pueden diagnosticar los lipomas basándose únicamente en la historia clínica y el examen físico, las pruebas de imagen pueden ser útiles en algunos casos.
Rayos X. Aunque estas pruebas crean imágenes claras de estructuras densas como el hueso, las radiografías simples pueden mostrar una sombra prominente causada por un tumor de tejido blando, como un lipoma.
Exploraciones de tomografía computarizada (TC). Estas exploraciones son más detalladas que las radiografías y, a menudo, mostrarán una masa grasa para confirmar el diagnóstico de lipoma.
Imágenes por resonancia magnética (IRM) . La mejor información para diagnosticar lipomas proviene de una resonancia magnética, que puede crear mejores imágenes de tejidos blandos como un lipoma. La resonancia magnética mostrará una masa grasa desde todas las perspectivas. A menudo, los médicos pueden hacer el diagnóstico de lipoma basándose únicamente en imágenes de resonancia magnética y no se requiere una biopsia.
Biopsia. A veces es necesaria una biopsia para confirmar el diagnóstico de lipoma. En una biopsia, se toma una muestra de tejido del tumor y se examina bajo un microscopio. Su médico puede administrarle un anestésico local para adormecer el área y tomar una muestra con una aguja. Las biopsias también se pueden realizar como una operación pequeña.
En la mayoría de los casos de lipoma, no es necesaria una biopsia para confirmar el diagnóstico. Después de extirpar el lipoma, se realizará una biopsia de una muestra del tejido.
Bajo un microscopio, los lipomas a menudo tienen una apariencia clásica con abundantes células grasas maduras. A veces también puede haber una pequeña cantidad de otros tipos de células, como cartílago o hueso.
Liposarcoma. Durante la fase de diagnóstico, su médico trabajará para diferenciar un lipoma de una forma más agresiva de tumor graso llamado liposarcoma. Los liposarcomas son cancerosos. Los síntomas del liposarcoma varían de los del lipoma. Los liposarcomas generalmente crecen rápidamente, a menudo son dolorosos y no son tan móviles como los lipomas.
Las personas con lipomas no tienen más probabilidades de desarrollar un cáncer graso en el futuro. La excepción son las personas con lipomas atípicos. Este subtipo de lipoma puede convertirse en un liposarcoma, pero esto es raro.
Debido a que los lipomas son tumores benignos, ningún tratamiento puede ser una opción, según sus síntomas. Si elige ningún tratamiento, es muy importante que consulte a su médico para realizar visitas periódicas para controlar cualquier cambio en el tumor.
El único tratamiento que eliminará por completo un lipoma es un procedimiento quirúrgico llamado escisión.
Procedimiento. En este procedimiento, generalmente se inyecta un anestésico local alrededor del tumor para adormecer el área. Los lipomas grandes o profundos pueden requerir anestesia regional o anestesia general. La anestesia regional adormece un área grande inyectando un medicamento anestésico en nervios específicos. La anestesia general lo pone a dormir.
Después de administrar la anestesia, su médico hará una incisión en su piel y extirpará el tumor.
Recuperación. Debería poder irse a casa poco después del procedimiento si se trata de una masa pequeña o superficial. Tendrá algunos puntos, que su médico retirará en un par de semanas.
El tiempo que tarde en volver a la mayoría de las actividades diarias dependerá del tamaño y la ubicación de su lipoma. Si tiene algún dolor o molestia, es posible que desee limitar alguna actividad. Su médico le dará instrucciones específicas para guiar su recuperación.
Reaparición. Los lipomas casi siempre se curan mediante una simple escisión. Es inusual que un lipoma vuelva a crecer pero, si vuelve a aparecer, la escisión es nuevamente la mejor opción de tratamiento.
Hay investigaciones en curso para obtener más información sobre los diversos subtipos de lipomas y por qué se forman en primer lugar. En el futuro, puede haber recomendaciones de tratamiento específicas para varios subtipos de lipoma.
Los cánceres más comunes que se diseminan a los huesos incluyen:
MBD puede causar dolor en el área de propagación y puede provocar daño y debilidad en el hueso. El hueso puede volverse tan débil que puede fracturarse con actividades de baja demanda o un trauma mínimo. Esto puede dificultar la participación incluso en actividades simples como caminar o estar de pie. Como resultado, la pérdida de calidad de vida es una gran preocupación para los pacientes con MBD.
Después del pulmón y el hígado, el esqueleto es el sitio más común de propagación de los cánceres que comienzan en los órganos. Las metástasis en el pulmón y el hígado a menudo no se detectan hasta una etapa avanzada de la enfermedad porque los pacientes no experimentan síntomas. Por el contrario, las metástasis óseas generalmente son dolorosas cuando ocurren.
El cáncer se disemina más comúnmente a estos sitios en el esqueleto:
A veces, el tumor conducirá a la destrucción del hueso en un área particular. Este proceso se denomina destrucción ósea osteolítica y es común en los cánceres que se han propagado a los huesos desde el pulmón, la tiroides, los riñones y el colon.
Alternativamente, se puede formar hueso nuevo en respuesta a la propagación del cáncer. Las nuevas lesiones formadoras de hueso u osteoblásticas hacen que el hueso se debilite y se deforme. Las lesiones osteoblásticas se observan con mayor frecuencia en la propagación del cáncer de próstata, vejiga y estómago.
El cáncer de mama a menudo se comporta de manera mixta osteolítica y osteoblástica. La enfermedad ósea metastásica osteolítica y osteoblástica se produce porque las diferentes células cancerosas secretan factores que interactúan con las células naturales del hueso y provocan la destrucción ósea, la formación de hueso nuevo o ambas cosas.
Debido a que MBD debilita los huesos afectados, las personas con esta enfermedad corren el riesgo de sufrir fracturas . Los huesos rotos causados por MBD se denominan fracturas patológicas.
A veces, el hueso aún no se ha roto, pero está tan débil que es inminente, lo que se conoce como fracturas patológicas inminentes. Los pacientes con fracturas inminentes o reales pueden verse obligados a permanecer en cama durante largos períodos de tiempo, lo que puede provocar posibles desequilibrios químicos en la sangre, como un aumento de los niveles de calcio (hipercalcemia). Hable con su médico acerca de las opciones para ayudar a prevenir las fracturas de huesos.
Los pacientes con cáncer que se ha propagado a los huesos de la columna pueden desarrollar daño en los nervios que puede provocar parálisis o pérdida del uso de las piernas y/o los brazos. Esto a menudo requerirá cirugía.
Un paciente con cáncer que experimente cualquier dolor, especialmente en la espalda, las piernas y los brazos, debe informar a su médico de inmediato. El dolor que ocurre sin actividad intensa (es decir, caminar o levantar un objeto) es especialmente preocupante.
Es importante que su médico comprenda su condición médica y sus síntomas. Su médico le hará preguntas sobre la naturaleza de cualquier dolor que tenga, su salud actual y sus condiciones médicas pasadas. La información que obtiene su médico se denomina historial médico.
Después de tomar su historial médico, su médico realizará un examen físico, concentrándose en las áreas dolorosas.
Rayos X. Después de la entrevista y el examen físico, su médico ordenará radiografías si sospecha que tiene MBD. Debido a que parte del dolor proviene de otras áreas (por ejemplo, experimentar dolor en la rodilla después de una lesión en la cadera), su médico puede ordenar radiografías del hueso más allá de las áreas donde siente molestias.
El examen de rayos X puede brindarle a un oncólogo una gran cantidad de información sobre si el hueso está afectado y cuánto.
Otras pruebas de imagen. Su médico también puede ordenar una gammagrafía ósea de todo el cuerpo. Esta prueba es útil para determinar si otros huesos, además del en cuestión, están involucrados con MBD. En casos selectos, su médico puede ordenar una tomografía computarizada (CT) y/o una imagen de resonancia magnética (MRI), especialmente si la columna vertebral o la pelvis pueden estar involucradas.
No se debe asumir un diagnóstico de enfermedad ósea metastásica a menos que un paciente tenga un cáncer primario conocido que se haya propagado previamente a los huesos.
Si tiene más de 45 años sin antecedentes personales de cáncer y una radiografía detecta un tumor óseo, debe programar una consulta con un oncólogo ortopédico (un cirujano ortopédico con capacitación avanzada en cirugía del cáncer).
El oncólogo ortopédico determinará si este tumor óseo es una metástasis de un cáncer primario desconocido o si es un cáncer óseo primario (sarcoma) .
Por ejemplo, si se ha destruido un área de hueso, los posibles diagnósticos incluyen enfermedad ósea metastásica o cáncer de hueso primario, como mieloma o linfoma. Los cánceres que comienzan en los huesos son mucho menos comunes en adultos mayores de 45 años. Otras posibles causas incluyen enfermedades como el sarcoma de Paget, el sarcoma posterior a la radiación, el hiperparatiroidismo y las fracturas por osteoporosis. Es probable que se necesiten pruebas adicionales para determinar el diagnóstico exacto.
Es posible que necesite una biopsia. Esto implica tomar un trozo de tejido del tumor y observarlo bajo un microscopio. Esto se puede hacer en una clínica a través de una biopsia con aguja o, más comúnmente, como un pequeño procedimiento quirúrgico.
La identificación temprana de MBD es muy importante. Cuanto más temprano sea el diagnóstico, más eficazmente los tratamientos pueden ayudar a mantener la función y la calidad de vida.
Los pacientes con cáncer deben ser evaluados de forma rutinaria con ciertas pruebas, que incluyen:
Después de que se completen los antecedentes, el examen físico, las radiografías y las pruebas de laboratorio, su médico determinará si tiene enfermedad ósea metastásica.
Es importante señalar que muchos pacientes con cáncer tienen dolor de huesos debido a ciertos tipos de quimioterapia, y que el hecho de que tenga dolor de huesos o articulaciones no significa que tenga MBD. Sin embargo, el seguimiento cuidadoso es muy importante si tiene dolor.
En muchos casos de enfermedad ósea metastásica, el cáncer ha progresado hasta el punto en que están involucrados múltiples sitios óseos. Como resultado, el tratamiento a menudo se enfoca en controlar los síntomas de dolor y debilidad ósea, y no pretende ser curativo.
Las opciones de tratamiento más comunes para MBD incluyen radiación, cirugía y medicamentos para controlar el dolor y prevenir una mayor propagación de la enfermedad. Su médico también puede recomendar una cirugía para estabilizar el hueso que está débil o roto.
Los pacientes con enfermedad ósea metastásica requieren un enfoque de equipo para la atención. Un oncólogo médico trabaja en estrecha colaboración con un cirujano ortopédico, que debe estar familiarizado con la enfermedad ósea metastásica, y un oncólogo radioterapeuta. Los especialistas en manejo del dolor y los trabajadores sociales también pueden formar parte de su equipo de tratamiento. Su médico oncólogo y/o cirujano determinará a cuál de estos proveedores debe consultar para las citas de seguimiento. Obtenga más información sobre las opciones de tratamiento para la enfermedad ósea metastásica para áreas específicas de propagación.
La radiación puede ser muy eficaz y es una de las terapias más utilizadas para tratar los síntomas en pacientes con MBD incurable. Al matar las células cancerosas, la radiación alivia el dolor, detiene el crecimiento del tumor y puede evitar que el hueso se quiebre. La radiación también se puede usar para controlar el cáncer después de la cirugía para reparar un hueso roto. La investigación muestra que la radiación después de la cirugía mejora la función del paciente y disminuye la necesidad de cirugías adicionales. Sin embargo, una vez que ocurre una fractura, la cirugía generalmente se realiza antes de la radiación.
MBD es un problema sistémico (en todo el cuerpo) y es poco probable que la radioterapia sea curativa. Antes del tratamiento, el médico y el paciente deben comprender claramente los objetivos de la radioterapia, ya sea para disminuir los síntomas y el dolor, o para destruir por completo la enfermedad en el hueso afectado.
Por lo tanto, el médico debe sopesar los posibles beneficios y riesgos de la radiación para cada paciente.
Diferentes tipos de cáncer responden de manera diferente a la radiación. Hay varios tipos de radioterapia disponibles.
Radiación de campo local. La radiación de campo local es el tipo más común de radiación que se usa para tratar la MBD. En este procedimiento, la radiación se dirige al tumor metastásico y al tejido adyacente inmediato. Segmentos óseos completos o múltiples huesos pueden ser objeto de radiación de campo local, dependiendo de cuántas áreas estén afectadas por la enfermedad.
El objetivo principal del tratamiento con radiación es aliviar el dolor con efectos secundarios mínimos. La radiación de campo local generalmente produce un alivio completo del dolor en 50 a 60 % de los casos y un alivio parcial en más del 80 % de los casos. La respuesta del MBD a la radiación depende de muchos factores, incluido el tipo de cáncer (por ejemplo, el cáncer de mama generalmente responde muy bien a la radiación, mientras que el cáncer de riñón no) y la ubicación del tumor.
El dolor generalmente comienza a disminuir en las primeras 1 a 2 semanas, pero el alivio máximo puede tardar varios meses. Por lo tanto, su médico le recetará analgésicos para que los tome durante el curso del tratamiento de radiación.
Irradiación de hemicuerpos. Esta radioterapia de campo grande a menudo se usa para pacientes con enfermedad metastásica generalizada. En lugar de enfocarse en huesos específicos, la irradiación del hemicuerpo se enfoca en áreas más grandes de la parte superior del cuerpo, la sección media o la parte inferior del cuerpo.
La mayoría de los pacientes con cáncer metastásico tienen múltiples tumores. La irradiación de hemicuerpos se usa para complementar la radiación de campo local y puede reducir la progresión de la enfermedad generalizada. Se utiliza con menos frecuencia que la radiación de campo local.
Terapia con radioisótopos. Una alternativa a la irradiación del hemicuerpo, la terapia con radioisótopos consiste en inyectar un medicamento radiactivo (radiofármaco) en una vena. Las áreas de enfermedad ósea metastásica absorben el radiofármaco, que luego mata las células tumorales. En comparación con la irradiación de hemicuerpos, la terapia con radioisótopos es más fácil de administrar a los pacientes y también es más fácil de tolerar para los pacientes.
Las opciones de tratamiento con medicamentos para pacientes con MBD incluyen:
La cirugía para MBD se usa para tratar o prevenir fracturas de huesos. Los objetivos son aliviar el dolor, reducir la necesidad de medicamentos para el dolor, restaurar la fuerza esquelética y recuperar la capacidad para realizar las actividades diarias.
Un hueso roto o debilitado debe fijarse cuidadosamente en su posición y sujetarse hasta que sea lo suficientemente fuerte para soportar el peso. Durante la cirugía, se puede extirpar el tumor y estabilizar el hueso con dispositivos de fijación, como alambres, placas, varillas, clavos y tornillos. A menudo, se coloca cemento óseo en el defecto creado por el tumor para darle mayor resistencia.
Las investigaciones muestran que a los pacientes que se someten a una cirugía para prevenir una rotura les va mucho mejor que a los que requieren cirugía después de que se produce una rotura.
La cirugía para reforzar los huesos con riesgo de fractura también permite que el médico oncólogo y el cirujano coordinen el tratamiento quirúrgico y la terapia sistémica.
La decisión de proceder con la cirugía es compleja e individualizada para cada paciente. Los cirujanos ortopédicos considerarán varios factores para determinar si un hueso está en riesgo de fracturarse. Estos incluyen si el sitio es doloroso, el tamaño del tumor y cómo se ve el hueso en una radiografía.
Debido a que los pacientes con enfermedad ósea metastásica generalmente son menos saludables que el paciente promedio que se somete a una cirugía ortopédica y la cirugía es más complicada, existe un aumento en los riesgos de rutina de la cirugía, que incluyen infección, sangrado, coágulos sanguíneos y daño a los nervios.
En consecuencia, el paciente, la familia, el cirujano y el oncólogo deben tomar una decisión cooperativa, informada y muy cuidadosa sobre si proceder con la cirugía.
Los avances en las técnicas quirúrgicas, las radioterapias y las terapias médicas han mejorado significativamente la calidad de vida de las personas que padecen cáncer que se ha diseminado al esqueleto desde su sitio de origen.
Las opciones de tratamiento para MBD se basan en cuánto se ha propagado el cáncer, qué huesos están afectados y la gravedad del daño óseo. Para obtener más información sobre las opciones de tratamiento para MBD en áreas específicas del esqueleto (como la parte superior del brazo o la pelvis): Enfermedad ósea metastásica: opciones de tratamiento para áreas específicas de propagación
Enfermedad ósea metastásica: opciones de tratamiento para áreas específicas de propagación
El veinte por ciento de las metástasis óseas ocurren en la extremidad superior (hombro, parte superior del brazo y antebrazo), y aproximadamente el 50% de las que ocurren en el húmero (parte superior del brazo). Las metástasis en las extremidades superiores pueden causar un deterioro funcional grave y pueden dificultar la higiene personal, la capacidad de moverse sin ayuda, la capacidad de utilizar ayudas externas (muletas, bastón, andador, etc.), el manejo de las comidas y las actividades generales de la vida diaria.
Las opciones de tratamiento incluyen:
La ubicación y extensión de la metástasis dicta la opción de tratamiento. Las lesiones metastásicas de la clavícula (clavícula) y el omóplato (escápula) generalmente se tratan sin cirugía. Algunos casos, sin embargo, requieren intervención quirúrgica.
El MBD del húmero superior cerca del hombro se puede tratar con una variedad de técnicas, según la extensión del cáncer. A veces, es necesario reemplazar una parte de la parte superior del brazo y el hombro con una prótesis metálica artificial (reemplazo protésico del húmero superior). En general, solo se reemplaza el lado del brazo de la articulación del hombro cuando un paciente tiene enfermedad metastásica. El lado de la cavidad de la articulación no suele estar afectado.
Estas cirugías son generalmente más complejas que los reemplazos de hombro que se usan para la artritis de hombro , y los pacientes con MBD a menudo tienen menos funciones debido a la extracción del manguito de los rotadores y la reconexión a la prótesis de metal.
Los tumores de la diáfisis humeral ocurren a lo largo del hueso, debajo del hombro y arriba del codo. También se tratan con una variedad de técnicas, aunque generalmente no es necesario reemplazar la articulación. El cemento óseo (polimetilmetacrilato o PMMA) proporciona una estabilidad inmediata, restaura la función y complementa la mala calidad del hueso. Las varillas humerales insertadas por el canal central del hueso abarcan todo el húmero y proporcionan estabilidad tanto mecánica como rotacional.
A veces, el tumor se extirpará si no es sensible a la radiación, pero a menudo el cirujano lo deja en su lugar porque el tratamiento con radiación puede destruir el tumor después de estabilizar el hueso.
Los espaciadores segmentarios (en los que se extrae la parte media del hueso y se reemplaza con metal) ofrecen una opción reconstructiva para el tratamiento de las lesiones de la diáfisis. Se utilizan en defectos grandes y en casos de cirugía previa fallida por progresión de la enfermedad. Los espaciadores segmentarios se pueden usar después de la resección de la lesión metastásica, lo que minimiza la pérdida de sangre en las lesiones sanguinolentas y, a menudo, alivia la necesidad de radiación posoperatoria.
La estabilización abierta con placas y tornillos es otra opción de tratamiento para las lesiones de la diáfisis humeral, aunque se utiliza con menos frecuencia que la fijación intramedular. La fijación abierta requiere una exposición más amplia del húmero y limita la capacidad de proteger todo el hueso.
Los tumores ubicados por encima del codo se pueden tratar con una variedad de técnicas. Los clavos flexibles ofrecen la capacidad de abarcar todo el húmero, una excelente recuperación funcional y la preservación de la articulación natural del codo. El reemplazo de codo puede ser necesario si el tumor se extiende a la articulación o involucra el extremo del húmero cerca de la articulación.
Las lesiones metastásicas debajo del codo son raras. Los tumores primarios más comunes que metastatizan a esta ubicación son el carcinoma de pulmón, mama y células renales (riñón). El cáncer de pulmón es el tumor primario más común que hace metástasis en la mano.
Las lesiones metastásicas en el radio y el cúbito se pueden tratar con varillas, placas y tornillos flexibles o aparatos ortopédicos.
El osteosarcoma es un tipo de cáncer en el que las células tumorales producen hueso inmaduro conocido como osteoide. Aunque el osteosarcoma es raro, es el cáncer de hueso infantil más común. Aproximadamente 1000 nuevos casos de osteosarcoma se diagnostican cada año en los Estados Unidos, aproximadamente 450 de estos son en niños y adolescentes.
El osteosarcoma generalmente afecta los huesos largos del cuerpo cerca de las placas de crecimiento , áreas donde se forma tejido nuevo a medida que crece una persona joven. La mayoría de los tumores ocurren alrededor de la rodilla, ya sea en el fémur (hueso del muslo) o en la tibia (espinilla). Sin embargo, el osteosarcoma también puede afectar otras partes del cuerpo e incluso puede surgir fuera de los huesos en los tejidos blandos, especialmente en pacientes de edad avanzada.
En algunos casos, el osteosarcoma puede hacer metástasis o diseminarse a otras áreas del cuerpo. Cuando los osteosarcomas se propagan, con mayor frecuencia se propagan a los pulmones.
Se desconoce la causa del osteosarcoma, pero los médicos han determinado una serie de factores que pueden hacer que una persona sea más propensa a desarrollar la enfermedad.
Los factores que pueden aumentar su riesgo de desarrollar osteosarcoma incluyen:
El dolor es el síntoma más común del osteosarcoma. Este dolor puede aparecer y desaparecer al principio pero, gradualmente, se vuelve constante. Algunos pacientes pueden tener hinchazón o una masa en el área del tumor. Si el tumor se encuentra en la pierna, el paciente también puede desarrollar cojera.
Debido a que el osteosarcoma debilita el hueso, algunos pacientes pueden desarrollar fracturas óseas patológicas en el área del tumor.
En raras ocasiones, los síntomas pueden ocurrir debido a la metástasis o diseminación del tumor a sitios distintos al hueso.
Su médico hablará con usted sobre su salud general y su historial médico y le preguntará acerca de sus síntomas. Él o ella luego examinará el área adolorida, en busca de:
Los estudios de imágenes y otras pruebas se utilizan para ayudar a diagnosticar el osteosarcoma.
Rayos X. Los rayos X brindan imágenes de estructuras densas, como el hueso, y son muy útiles para diagnosticar el osteosarcoma. El osteosarcoma puede aparecer de diferentes maneras en una radiografía, pero los hallazgos típicos incluyen:
Si bien muchos osteosarcomas parecen osteoblásticos (lo que significa que producen hueso nuevo) en las radiografías, los hallazgos menos comunes incluyen lesiones que parecen osteolíticas (lo que significa que rompen el hueso) u osteolíticas/osteoblásticas mixtas. Las lesiones osteoblásticas aparecen blancas en la radiografía, mientras que las lesiones osteolíticas aparecen oscuras/negras.
Imágenes por resonancia magnética (IRM). Estos estudios proporcionan vistas detalladas de la médula ósea y otros tejidos blandos. Una resonancia magnética ayudará a su médico a determinar la extensión del tumor y buscar la propagación del cáncer a otras partes de su cuerpo.
Exploración por tomografía computarizada (TC). Estos estudios proporcionan imágenes transversales que pueden ayudar a su médico a evaluar más a fondo el tumor. Si bien las resonancias magnéticas generalmente se consideran más útiles que las tomografías computarizadas en el estudio de los osteosarcomas, se puede ordenar una tomografía computarizada para pacientes que no pueden tolerar una resonancia magnética cerrada.
Pruebas de laboratorio. Su médico puede ordenar una serie de pruebas de laboratorio, incluida una prueba de sangre llamada fosfatasa alcalina sérica . Es importante tener en cuenta que, aunque un nivel elevado de fosfatasa alcalina puede indicar un diagnóstico de osteosarcoma, también puede ocurrir por otras razones. Por ejemplo, existe una amplia gama de niveles normales de fosfatasa alcalina en niños y adolescentes en crecimiento, una gran parte de la población afectada por osteosarcoma. Además, a veces se puede observar un nivel elevado de fosfatasa alcalina en otras enfermedades que involucran el crecimiento óseo.
A pesar de estas limitaciones, su médico puede ordenar la prueba para obtener una mejor imagen general de su condición y porque se sabe que los niveles de fosfatasa alcalina en el osteosarcoma están relacionados con el pronóstico.
Biopsia _ Se necesita una biopsia para confirmar el diagnóstico de osteosarcoma. En una biopsia, se extrae una pequeña muestra del tumor y se examina bajo un microscopio. El procedimiento se puede realizar bajo anestesia local con una aguja o como una pequeña operación abierta.
Durante la biopsia, su médico buscará osteoide irregular y otros rasgos característicos de los osteosarcomas. El osteoide irregular a menudo aparece en una colección de células que tienen una apariencia en forma de huso.
calificación El tumor será “graduado” durante la biopsia. En general, cuanto más alto es el grado, más agresivo se sospecha que es el tumor.
Los tumores de grado bajo (menos agresivos) generalmente se parecen más a las células normales que los tumores de grado intermedio o alto. Los tumores de alto grado (más agresivos) no se parecen en nada al tejido circundante y, a menudo, se asocian con un peor pronóstico.
Puesta en escena. Una vez que el tumor se haya identificado como osteosarcoma, su médico usará estudios por imágenes y otras pruebas para determinar si el cáncer se ha propagado. Este proceso se conoce como “puesta en escena”. Identificar la etapa del tumor ayudará a su médico a determinar la estrategia de tratamiento más efectiva.
Los médicos han identificado varios subtipos diferentes de osteosarcoma, según el aspecto de los tumores en las radiografías y bajo el microscopio. El subtipo específico de osteosarcoma se determina durante la biopsia. Es importante saber qué subtipo tienes, ya que cada uno progresa de forma diferente y responde de forma diferente a determinados tratamientos.
Algunos de los subtipos más comunes se describen a continuación.
Osteosarcomas convencionales. Este es el subtipo más común de osteosarcoma. Los osteosarcomas convencionales ocurren con mayor frecuencia cerca de la rodilla, en el extremo inferior del fémur (hueso del muslo) o en el extremo superior de la tibia (espinilla).
Los osteosarcomas convencionales se dividen en tres tipos diferentes, según su composición:
Osteosarcoma telangiectásico. Los osteosarcomas telangiectásicos son tumores vasculares que producen una pequeña cantidad de osteoide. En la radiografía, aparecen como lesiones destructivas que a veces pueden confundirse con otros tipos de tumores. Cuando se observan a simple vista, los osteosarcomas telangiectásicos tienen una apariencia distintiva y, a menudo, los médicos los describen como “una bolsa de sangre”.
Osteosarcoma de células pequeñas. Cuando se examina bajo un microscopio, el osteosarcoma de células pequeñas se compone de pequeñas células azules redondas. Los tumores tienen una apariencia similar al sarcoma de Ewing, otro tipo de cáncer de hueso. Para diferenciar entre los dos tipos de cáncer, su médico buscará la producción de osteoide, una característica de los osteosarcomas.
Osteosarcoma superficial de alto grado. Estos tumores surgen de la superficie del hueso. Aunque hay otros osteosarcomas que surgen de la superficie del hueso (tumores paranasales y periósticos), las células en los osteosarcomas de superficie de alto grado tienen un aspecto más irregular.
Osteosarcoma de Paget. Este subtipo ocurre en pacientes con enfermedad ósea de Paget, una afección ósea crónica que ocurre con mayor frecuencia en pacientes mayores de 60 años.
Osteosarcoma posradiación. La exposición previa a la radiación en el tratamiento de otros tipos de cáncer puede provocar osteosarcoma posterior a la radiación. Esto es especialmente cierto si la dosis de radiación fue alta.
Osteosarcoma extraesquelético. Estos tumores surgen en los tejidos blandos del cuerpo, en lugar de en los huesos. El osteosarcoma extraesquelético es más común en adultos mayores.
Osteosarcoma perióstico. Los osteosarcomas periósticos surgen de la superficie del hueso, con mayor frecuencia en la diáfisis, o parte del eje, de la tibia (espinilla). Los tumores producen cartílago y osteoide.
Osteosarcoma central de bajo grado. Estos tumores ocurren en el eje de los huesos largos del cuerpo. Por lo general, progresan lentamente, pero pueden reaparecer como tumores de alto grado más agresivos que tienen un mayor riesgo de diseminación.
Osteosarcoma parostal. Los osteosarcomas para óseos surgen en la superficie de los huesos largos y, con mayor frecuencia, se presentan como una masa dura en la parte posterior del fémur inferior (hueso del muslo), detrás de la rodilla.
El tratamiento del osteosarcoma depende de varios factores, entre ellos:
Al tratar el osteosarcoma, el médico trabajará en estrecha colaboración con el paciente y su familia para desarrollar el mejor plan de tratamiento posible.
Aunque el tratamiento es individualizado, muchos osteosarcomas se tratan de manera similar con una combinación de quimioterapia y cirugía.
La quimioterapia es el uso de medicamentos para tratar el cáncer. En pacientes con osteosarcoma, la quimioterapia se administra antes de la cirugía para ayudar a reducir el tamaño del tumor y prevenir la propagación del cáncer. Luego se vuelve a administrar, después de la cirugía, para atacar cualquier célula cancerosa que pueda quedar y reducir el riesgo de que el cáncer regrese.
Se utilizan varios medicamentos en el tratamiento de quimioterapia para el osteosarcoma. Los más comunes incluyen doxorrubicina (DOX), metotrexato (MTX), dactinomicina (BCD), ciclofosfamida (INN) y cisplatino (CDDP). La mayoría de los pacientes reciben dos o más de estos medicamentos a la vez, lo que se conoce como quimioterapia combinada.
El papel de la quimioterapia en el tratamiento de los osteosarcomas parósticos y periósticos es un tema de investigación actual. Dado que muchos de estos tumores son de grado bajo o intermedio y no han invadido gran parte del tejido circundante, a menudo se pueden tratar solo con cirugía. Sin embargo, los osteosarcomas parósticos y periósticos más agresivos a menudo requieren quimioterapia.
Actualmente, el tratamiento quirúrgico del osteosarcoma localizado implica con mayor frecuencia una cirugía de salvamento de la extremidad en lugar de una amputación. En la cirugía de salvamento de una extremidad, el médico extirpa el tumor y una parte del tejido sano que lo rodea, mientras conserva la mayor cantidad posible de tendones, nervios y vasos sanguíneos cercanos. La sección de hueso que se extrae se reemplaza con un injerto óseo (hueso tomado de un donante o de otra parte de su propio cuerpo) o una prótesis hecha de metal y otros materiales.
Cuando se extirpa el tumor, se analizará bajo un microscopio para garantizar la extirpación completa y determinar la respuesta de los tejidos a la quimioterapia preoperatoria. Si se determina que el tumor no se extirpó por completo, es posible que se necesite otra cirugía para extirpar el resto. Si la respuesta del tejido a la quimioterapia preoperatoria fue deficiente (lo que significa que no se eliminó una cantidad suficiente de células tumorales), su médico puede recomendar diferentes medicamentos de quimioterapia después de la cirugía. Si la respuesta del tejido a la quimioterapia preoperatoria fue buena, su médico generalmente recomendará continuar con el mismo régimen de quimioterapia que antes.
La radioterapia utiliza dosis altas de rayos X para eliminar las células cancerosas y reducir los tumores. Aunque la radioterapia no se usa comúnmente para tratar el osteosarcoma, se puede recomendar para ciertos pacientes.
La recuperación después de la cirugía de salvamento de extremidades requiere una extensa fisioterapia, rehabilitación y atención de seguimiento. Los niños, especialmente, necesitan ser alentados y apoyados por familiares y amigos durante este tiempo. Mientras continúan con la quimioterapia, pueden tener un nivel de energía disminuido y carecer de motivación para hacer ejercicio.
Se necesitarán visitas de seguimiento periódicas después del tratamiento para detectar complicaciones del tratamiento y la recurrencia o propagación del cáncer. Estas visitas pueden incluir análisis de sangre o estudios por imágenes, como radiografías o tomografías computarizadas.
El pronóstico del paciente después del tratamiento dependerá de muchos factores, entre ellos:
Sin embargo, en la mayoría de los casos, la respuesta del tumor a la quimioterapia preoperatoria es el mejor predictor del pronóstico. En el momento de extirpar el tumor, el médico lo examinará bajo un microscopio para determinar la cantidad de necrosis (cuánto tumor ha muerto). En general, cuanto mayor sea el porcentaje de necrosis, mejor será el pronóstico.
El tratamiento es exitoso para muchos pacientes con osteosarcoma. Sin embargo, debido a que cada situación es única, es mejor hablar con su médico sobre el pronóstico específico para usted o su hijo.
Este sarcoma raro de bajo grado fue identificado por Meis-Kindblom y Kindblom en 1998.Los pacientes están en su edad adulta media (rango de tercera a novena década, la mediana de 53 años) y los hombres y las mujeres son igualmente afectados.El tumor se presenta como una masa indolora de meses o años de duración. Las lesiones promedio de 3 cm de tamaño.Examen de resonancia magnética demostrará la extensión del tumor y los ganglios linfáticos regionales y el pecho debe ser evaluado.Se requiere escisión con un margen amplio y largo plazo de seguimiento.
Información completa sobre este tumor
Introducción y Definición: Este sarcoma rara de bajo grado fue identificado por Meis-Kindblom y Kindblom en 1998. Aproximadamente el 30% de estos tumores se producen en el pie y el tobillo, el 65% de la mano y la muñeca, y el resto en otros sitios. Las lesiones promedio de 3 cm de tamaño. La masa es mal delimitada y subcutánea, lo que lleva a un mal diagnóstico frecuente como quiste ganglionar o tenosinovitis.
Incidencia y Demografía: Los pacientes están en su edad adulta media (rango de tercera a novena década, la mediana de 53 años) y los hombres y las mujeres son igualmente afectados.
Síntomas y Presentación: El tumor se presenta como una masa indolora de meses o años de duración.X-Ray Apariencia y Advanced Imaging
Hallazgos: Examen de resonancia magnética demostrará la extensión del tumor y los ganglios linfáticos regionales y el pecho debe ser evaluado.
Opciones de tratamiento para este tumor: El manejo adecuado comienza con el reconocimiento del potencial de malignidad de masas aparentemente inocentes. Se requiere escisión con un margen amplio y largo plazo de seguimiento.Resultados del
Tratamiento y pronóstico: Debido a la presentación inocua los márgenes quirúrgicos iniciales son a menudo insuficientes. La recurrencia se observa en 60 a 70 por ciento de los casos. Aproximadamente una cuarta parte de los casos requieren amputación debido a la recurrencia local de recurrente. La metástasis es poco frecuente, pero la diseminación del tumor a los ganglios linfáticos y el pulmón regionales han sido reportados.El pronóstico es similar a otros sarcomas de bajo grado, con dos tercios de los pacientes vivos sin enfermedad y el resto vivo con la enfermedad a los cinco años de seguimiento.
Adamantinoma
Adamantinoma de los huesos largos, es de crecimiento lento, de bajo grado y extremadamente raro, maligno de origen epitelial, que se produce casi exclusivamente en la tibia y el peroné.
El tumor generalmente se presenta en la segunda a la quinta década de la vida.
El paciente suele tener hinchazón que puede ser doloroso. La duración de los síntomas puede variar desde unas pocas semanas hasta años.
Adamantinoma aparece como una lesión excéntrica, bien circunscrita, y lítico en la radiografía simple.
Adamantinoma se trata mediante la extirpación quirúrgica amplia
Información completa sobre este tumor
Introducción y Definición:
Adamantinoma de los huesos largos, o adamantinoma extragnathic, es un bajo grado del tumor extremadamente raro, malignas de origen epitelial. No está relacionado a adamantinoma o ameloblastoma de la mandíbula y el maxilar que se deriva de la bolsa de Rathke. Adamantinoma es un tumor localmente agresivo osteolítica
Incidencia y Demografía:
El tumor generalmente se presenta en la segunda a la quinta década de la vida, pero puede afectar a los pacientes entre las edades de 3 a 73. La mayoría de los pacientes son hombres.
Síntomas y Presentación:
El sitio de 90% del tiempo en la diáfisis de la tibia con las lesiones restantes se encuentran en el peroné y los huesos tubulares largos. A menudo hay una historia de trauma asociado con adamantinoma pero su papel en el desarrollo de esta lesión no está claro. El paciente suele tener hinchazón que puede ser doloroso. La duración de los síntomas puede variar desde unas pocas semanas hasta años.
X-Ray Apariencia y Advanced Imaging Hallazgos:
Adamantinoma aparece como una lesión excéntrica, bien circunscrita, y lítico en la radiografía simple. La corteza anterior de la tibia es, con mucho, el lugar más común. La lesión suele tener varios defectos líticos separadas por hueso esclerótico que le da una apariencia de “pompa de jabón”. Hay adelgazamiento cortical pero poca reacción perióstica. La lesión puede romper a través de la corteza y se extienden en el tejido blando. Puede haber múltiples lesiones adyacentes con hueso intervenir normal. RM ayuda a demostrar la intraósea y la participación extraósea. El radiológicamente diagnóstico diferencial incluye la displasia osteofibrous, displasia fibrosa, ABC, fibroma condromixoide y el condrosarcoma
Diagnóstico diferencial:
El radiológicamente diagnóstico diferencial incluye la displasia osteofibrosa, displasia fibrosa, ABC, fibroma condromixoide y el condrosarcoma.
Técnica de biopsia preferida para este tumor:
Biopsia abierta
Hallazgos histopatológicos:
En el examen macroscópico, adamantinoma está bien demarcada y lobulada. El tumor de color gris o blanco es correoso y puede tener áreas focales de hemorragia y necrosis. Espículas óseas y quistes llenos de líquido en la sangre o de color pajizo también puede ser present.Adamantinoma es un tumor bifásico con islas de células epiteliales rodeadas por un estroma fibroso reactivo soso. El estroma se compone de células productoras de colágeno en forma de huso. Los nidos de células malignas son columnares y tener una empalizada periférica. Diferenciación escamosa y la producción de queratina son raros. El tumor es positivo en la tinción inmunohistoquímica con el anticuerpo queratina. El origen epitelial se confirma cuando las membranas basales, los desmosomas y los filamentos toneladas son vistos bajo el microscopio electrónico.
Opciones de tratamiento para este tumor:
Adamantinoma se trata mediante la extirpación quirúrgica amplia. Este tumor es insensible a la radiación y puede producir metástasis en los pulmones, ganglios linfáticos y órganos abdominales por ambas rutas hematógenas y linfático. No se utiliza la quimioterapia.
Margen de Preferencia para este tumor:
Ancho
Resultados del Tratamiento y pronóstico:
En el 20% de los casos hay metástasis tardías en el curso de la enfermedad.
Características especiales y Inusuales
Displasia osteofibrosa, o fibroma osificante, es otra lesión con una predilección llamativa para la tibia que tiene una asociación bien documentado con adamantinoma y puede ser un precursor benigno a ella.
Lecturas recomendadas y Referencia:
Conway, WF y CW Hayes, Varios lesiones del hueso, radiológicos Clinics of North America, 31 (2): 339357, de marzo de 1993. Bulloughs, Andrew, ortopédica Pathologv (tercera edición), Times Mirror Internacional Publishers Limited, Londres, 1997 Huvos , Andew. Hueso Tumors- Diagnóstico. Tratamiento y pronóstico, WBSaunders, Co., 1991. Fletcher, Christopher. Histopatología de diagnóstico de tumores, Churchill Livingstone, 1995. Van Der Woude, HJ AJR Am J Roentgenol. 2004 Dec; 183 (6): 1737-
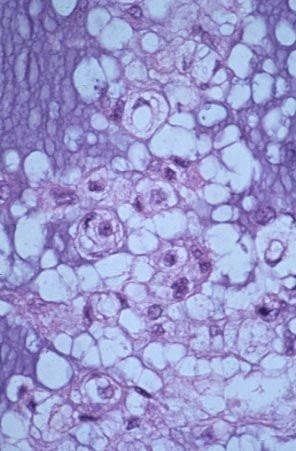
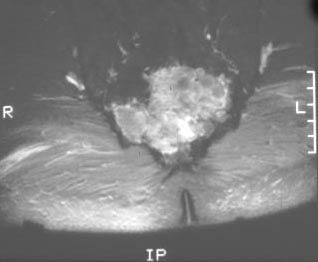
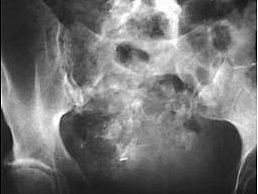
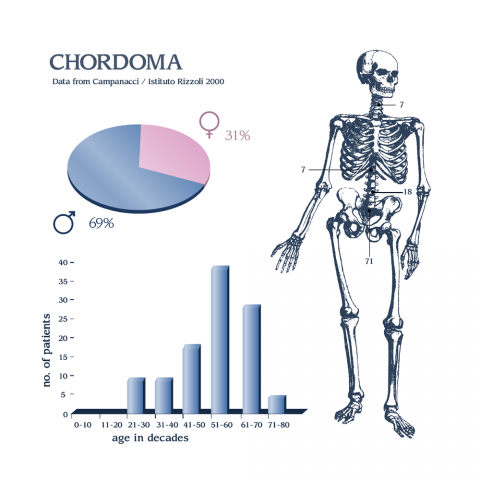
Diafisaria estenosis medular con histiocitoma fibroso maligno (síndrome de Hardcastle) es una displasia rara, autosómica dominante, caracterizada por huesos simétricos estenosis medular diafisaria de los huesos largos y un riesgo muy elevado de desarrollo de histiocitoma fibroso maligno.
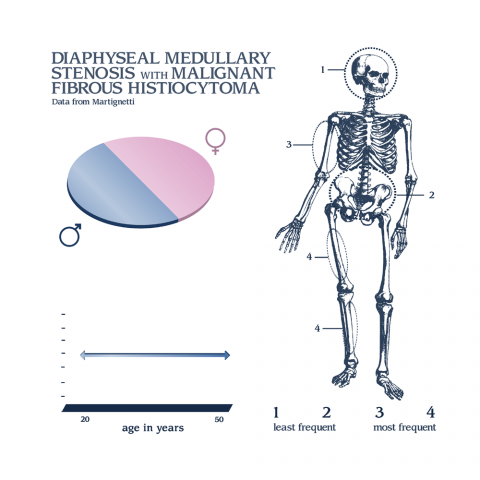
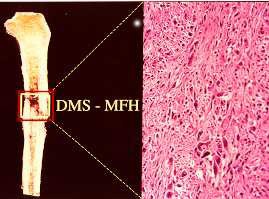
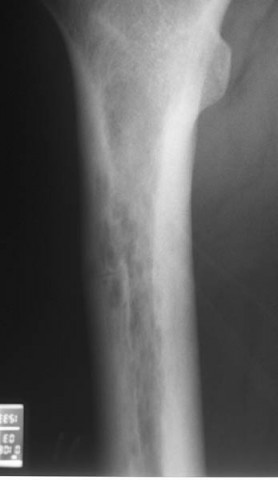
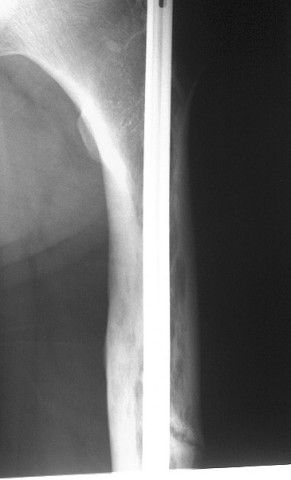
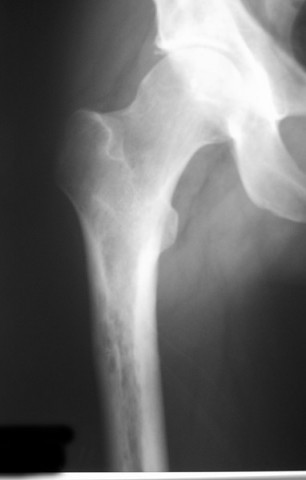
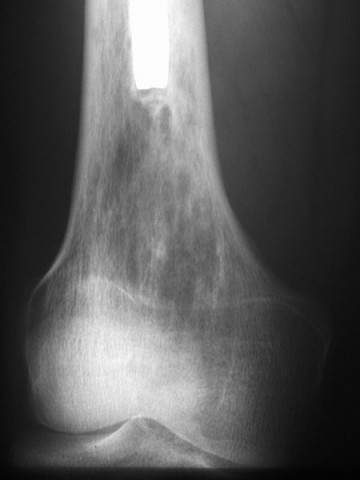
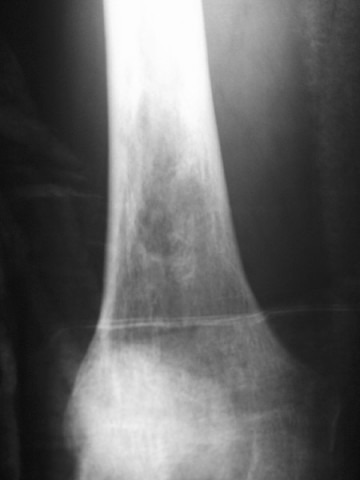
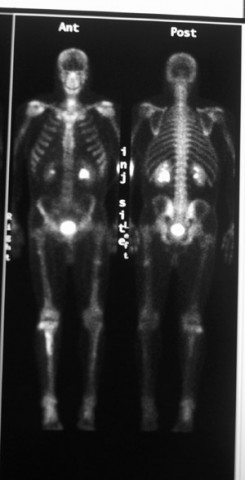
El síndrome de Hardcastle es una displasia rara, autosómica dominante, caracterizada por huesos simétricos estenosis medular diafisaria de los huesos largos y un riesgo muy elevado de desarrollo de histiocitoma fibroso maligno.
Los autores de este sitio, en colaboración con el Dr. Martignetti, tienen interés en este trastorno. Si usted o un miembro de su familia se ve afectada por este trastorno, por favor póngase en contacto con los autores de este sitio.
Esta rara enfermedad ha sido reportada en cuatro o cinco familias en todo el mundo. La enfermedad se hereda como un rasgo autosómico dominante. Los hallazgos característicos son visibles en las radiografías en la edad adulta temprana. Transformación en histiocitoma fibroso maligno se presenta en aproximadamente el 35 por ciento. Insuficiencia fracturas pueden ocurrir en los países afectados los huesos largos. Después del tratamiento, la curación es lento y puede dar lugar a falta de unión.
Los autores tienen experiencia con las personas afectadas a partir de dos de las cinco familias conocidas. Una persona ha sufrido fracturas traumáticas del fémur y la tibia. Si bien el hueso afectado parece muy densa en las radiografías simples, el hueso es más probable que la fractura de un hueso normal.
Diafisaria estenosis medular con histiocitoma fibroso maligno (síndrome de Hardcastle) es una displasia rara, autosómica dominante, caracterizada por huesos simétricos estenosis medular diafisaria de los huesos largos y un riesgo muy elevado de desarrollo de histiocitoma fibroso maligno.
El síndrome de Hardcastle es una displasia rara, autosómica dominante, caracterizada por huesos simétricos estenosis medular diafisaria de los huesos largos y un riesgo muy elevado de desarrollo de histiocitoma fibroso maligno.
Los autores de este sitio, en colaboración con el Dr. Martignetti, tienen interés en este trastorno. Si usted o un miembro de su familia se ve afectada por este trastorno, por favor póngase en contacto con los autores de este sitio.
Esta rara enfermedad ha sido reportada en cuatro o cinco familias en todo el mundo. La enfermedad se hereda como un rasgo autosómico dominante. Los hallazgos característicos son visibles en las radiografías en la edad adulta temprana. Transformación en histiocitoma fibroso maligno se presenta en aproximadamente el 35 por ciento. Insuficiencia fracturas pueden ocurrir en los países afectados los huesos largos. Después del tratamiento, la curación es lento y puede dar lugar a falta de unión.
Los autores tienen experiencia con las personas afectadas a partir de dos de las cinco familias conocidas. Una persona ha sufrido fracturas traumáticas del fémur y la tibia. Si bien el hueso afectado parece muy densa en las radiografías simples, el hueso es más probable que la fractura de un hueso normal.
Los hemangiomas son lesiones benignas del hueso caracterizada por espacios vasculares revestidos con células endoteliales. Aproximadamente el 50% de los hemangiomas óseos se encuentran en los cuerpos vertebrales (torácicos especialmente) y el 20% se encuentran en la bóveda craneal. Las lesiones restantes se encuentran en la tibia, fémur y el húmero.
La aparición en X-ray depende de la ubicación. Las lesiones son a menudo mal definido, que aparece como un área algo localizada de vasos abundante, dilatada, algunas de ellas pueden estar en la superficie del hueso, dentro de la corteza del hueso, o extenderse a la cavidad medular.
Los hemangiomas son lesiones asintomáticas generalmente descubierto en una radiografía o una autopsia. hemangiomas vertebrales pueden causar síntomas neurológicos si se extienden en el espacio epidural. Los síntomas pueden variar con otros factores que causan la distensión vascular o de reacción, tales como la dependencia, la actividad, el embarazo y la menstruación.
| Caso | Síntomas y presentación |
|---|---|
| Adenoma / adenocarcinoma papilar digital agresivo – Pie y tobillo | Puede tener un curso clínico indolente sin síntomas ni cambios durante años. |
| Sarcoma fibroblástico mixoinflamatorio acral – Pie y tobillo | El tumor se presenta como una masa subcutánea indolora de meses o años de duración. |
| Sarcoma fibroblástico mixoinflamatorio acral – Pie y tobillo | El tumor se presenta como una masa subcutánea indolora de meses o años de duración. |
| Adamantinoma | El sitio el 90% del tiempo en la diáfisis de la tibia con las lesiones restantes se encuentran en el peroné y los huesos tubulares largos. A menudo hay antecedentes de traumatismo asociado con el adamantinoma, pero su papel en el desarrollo de esta lesión sigue sin estar claro. El paciente suele presentar hinchazón que puede ser dolorosa. La duración de los síntomas puede variar desde unas pocas semanas hasta años. |
| Adamantinoma | El sitio el 90% del tiempo en la diáfisis de la tibia con las lesiones restantes se encuentran en el peroné y los huesos tubulares largos. A menudo hay antecedentes de traumatismo asociado con el adamantinoma, pero su papel en el desarrollo de esta lesión sigue sin estar claro. El paciente suele presentar hinchazón que puede ser dolorosa. La duración de los síntomas puede variar desde unas pocas semanas hasta años. |
| Fibromatosis agresiva (tumor desmoide) | |
| Adenoma / adenocarcinoma papilar digital agresivo – Pie y tobillo | Puede tener un curso clínico indolente sin síntomas ni cambios durante años. |
| Un niño de 9 años tiene dolor en la rodilla derecha después de caerse durante un partido de fútbol. | Un niño de 9 años tiene dolor en la rodilla derecha después de caerse durante un partido de fútbol. Las radiografías se muestran en las Figuras 13a y 13b. La lesión ocurrió hace dos semanas y el dolor ha mejorado significativamente. ¿Cuál es el siguiente paso más apropiado en la atención de este paciente? |
| Quiste óseo aneurismático: pie y tobillo | Puede haber antecedentes de trauma y algunos han postulado un vínculo causal entre el trauma y esta lesión. Los pacientes se quejan de dolor y una masa de crecimiento lento. |
| Quiste óseo aneurismático | Dolor que aumenta gradualmente, una masa o con una fractura patológica a través de la lesión. En unos pocos casos se ha informado de un rápido aumento del tamaño de la lesión. |
| Quiste óseo aneurismático: pie y tobillo | Puede haber antecedentes de trauma y algunos han postulado un vínculo causal entre el trauma y esta lesión. Los pacientes se quejan de dolor y una masa de crecimiento lento. |
| Angiosarcoma | El angiosarcoma de alto grado parece tener dos presentaciones clínicas distintas. Primero, la lesión puede presentarse como múltiples lesiones en un solo hueso, dos o más huesos adyacentes o quizás todos los huesos de una extremidad. Estas lesiones parecen tener un curso indolente y el pronóstico sigue siendo bueno. La segunda presentación es la de lesiones únicas o múltiples de rápida progresión que hacen metástasis a otros huesos o al pulmón, esta forma de la enfermedad tiene un pronóstico muy precario. Este caso ilustró el tipo posterior. |
| Angiosarcoma | El angiosarcoma de alto grado parece tener dos presentaciones clínicas distintas. Primero, la lesión puede presentarse como múltiples lesiones en un solo hueso, dos o más huesos adyacentes o quizás todos los huesos de una extremidad. Estas lesiones parecen tener un curso indolente y el pronóstico sigue siendo bueno. La segunda presentación es la de lesiones únicas o múltiples de rápida progresión que hacen metástasis a otros huesos o al pulmón, esta forma de la enfermedad tiene un pronóstico muy precario. Este caso ilustró el tipo posterior. |
| Irregularidad cortical avulsiva – “lesión por tirón” | Los pacientes se presentan con dolor de rodilla y lesión ósea. Uno o ambos lados pueden ser sintomáticos. |
| Mixoma celular benigno | Una masa palpable con dolor local o una masa indolora de crecimiento lento. |
| Histiocitoma fibroso benigno | Clínicamente, los pacientes refieren dolor por la lesión, a menudo de meses o años de duración. El dolor puede estar asociado con una fractura patológica. Puede haber algo de sensibilidad local, pero no se observa hinchazón o masa, y no hay síntomas sistémicos. Normalmente no hay deterioro de la función de la articulación cercana. Las lesiones de la columna pueden causar un defecto neurológico al presionar la médula espinal. En algunos casos, existe un trastorno subyacente primario del metabolismo del colesterol u otras anomalías de los lípidos. En estos casos, las lesiones óseas líticas son análogas a las observadas en enfermedades de almacenamiento como la enfermedad de Gaucher. Estas múltiples lesiones se denominan “xantoma diseminado”. Un caso reportado es el de un niño de 10 años con lesiones líticas en pelvis, fémur y húmero, así como pápulas y placas amarillas y marrones en cara y tronco. Este paciente también tenía poliuria y polidipsia, y se encontró que tenía diabetes insípida. (Khandpur) Radiográficamente, la lesión ocurre comúnmente en las costillas, la pelvis, incluidos el sacro y el ilion, o en la epífisis o diáfisis de los huesos tubulares. Estos tumores se han informado en la mandíbula y tejidos blandos asociados. En otro informe, este tumor se presentó comúnmente alrededor de la rodilla. |
| Extraña proliferación osteocondromatosa parosteal | Hay antecedentes de una masa levemente dolorosa que parece aumentar de tamaño durante muchas semanas o meses. Por lo general, no hay antecedentes de trauma. Puede haber rigidez de una articulación cercana u otros síntomas mecánicos. |
| Extraña proliferación osteocondromatosa parosteal | Hay antecedentes de una masa levemente dolorosa que parece aumentar de tamaño durante muchas semanas o meses. Por lo general, no hay antecedentes de trauma. Puede haber rigidez de una articulación cercana u otros síntomas mecánicos. |
| Isla de huesos | |
| Condrosarcoma de células claras | El tumor es una lesión de crecimiento lento que tiene predilección por la epífisis de los huesos largos principales. Por lo tanto, la aparición gradual de dolor relacionado con las articulaciones en la cadera o el hombro es una presentación típica. A veces, una fractura patológica obligará al paciente a ver tratamiento para la lesión, a pesar de que ha habido un dolor leve o moderado durante años. La presentación general de esta lesión imita la de un tumor óseo benigno. |
| Caso | Síntomas y presentación | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor marrón | Clínicamente, el hiperparatiroidismo se presenta como “piedras, huesos y gemidos”. Los cálculos se refieren a cálculos renales recurrentes. Huesos se refiere a las lesiones óseas que ocurren en casos severos o prolongados. Groans está destinado a describir los síntomas gastrointestinales de náuseas, vómitos, úlceras pépticas y pancreatitis, así como la obnubilación que ocurre con la hipercalcemia. | ||||||||||
| Periartritis calcificada | Los pacientes se presentan con dolor local, sensibilidad, hinchazón y calor. | ||||||||||
| Cáncer de mama metastásico | El dolor es el síntoma de presentación más común. La fractura patológica rara vez ocurre sin antecedentes de unas pocas semanas o meses de dolor cada vez más intenso. En algunos casos, el paciente ha intentado ignorar o negar los síntomas. A veces, se piensa que una lesión ósea dolorosa es un “tirón muscular” o un “esguince” y se prescriben analgésicos fuertes, lo que permite que el paciente continúe tolerando un dolor muy intenso antes de que se descubra la verdadera naturaleza del problema. También pueden aparecer síntomas sistémicos, como hipercalcemia. | ||||||||||
| Cáncer de próstata metastásico | El diagnóstico de cáncer de próstata generalmente comienza con PSA sérico elevado y / o fosfatasa ácida prostática, o la palpación de nódulos en un tacto rectal. El diagnóstico generalmente se confirma mediante una biopsia guiada por ecografía transrectal. El PSA también es actualmente el marcador más útil para evaluar el nivel de afectación ósea en el cáncer de próstata. El diagnóstico de metástasis óseas se realiza mediante una gammagrafía ósea. El problema con las gammagrafías óseas es que se consideran sensibles en el diagnóstico inicial, pero la especificidad es relativamente baja. Tampoco son adecuados para controlar la respuesta al tratamiento a corto plazo. Actualmente se están investigando otros marcadores para medir la cantidad de afectación ósea. Uno de estos marcadores, el telopéptido carboxi-terminal reticulado de piridinolina de colágeno tipo I (ICTP), se escinde durante la degradación del colágeno tipo I, lo que es indicativo de recambio óseo. El pensamiento actual es que el ICTP es más específico para el hueso que el PSA, pero es necesario realizar más estudios.
Se desconoce el mecanismo real por el cual las células cancerosas metastásicas provocan reacciones osteoblásticas. Sin embargo, Koeneman et al han propuesto que “para prosperar en el entorno óseo, las células cancerosas deben adquirir propiedades ‘similares a las de las células óseas'”. Se cree que existen interacciones celulares recíprocas entre las células del cáncer de próstata y el estroma óseo que dan como resultado la proliferación tanto del cáncer de próstata como de las células del estroma óseo. Esta interacción implica la participación de factores de crecimiento, como TGF-ß, bFGF e IGF tanto en el hueso como en la próstata, que se modula además por las acciones diferenciadoras de las proteínas morfogenéticas óseas y las proteínas relacionadas con la PTH. Al final, las condiciones son adecuadas para el crecimiento osteoblástico y las consecuencias son dolor óseo, inflamación y un mayor riesgo de fracturas. Los tumores vertebrales pueden comprimir la médula espinal y causar daño a los nervios. Es importante tener una buena idea del nivel de afectación ósea, ya que tiene un impacto significativo en la supervivencia general del paciente. Los métodos actuales de tratamiento para el cáncer de próstata son la prostatectomía, la radioterapia y la terapia hormonal. La terapia hormonal se puede lograr mediante algunos métodos diferentes. La orquiectomía y la administración de estrógenos son dos opciones. Aunque el 60-80% de los pacientes con cáncer de próstata avanzado mejoran después de la castración, hay una progresión inevitable a un estado independiente de andrógenos testicular, en el que los andrógenos suprarrenales se hacen cargo. También existen fármacos que afectan directamente a la glándula pituitaria, como los agonistas de LHRH, y algunos pacientes también reciben fármacos antiandrógenos para bloquear el efecto de los andrógenos producidos por las glándulas suprarrenales produciendo un bloqueo total de andrógenos. Algunos pacientes con metástasis a menudo requieren un tratamiento adicional, como la terapia hormonal de segunda línea, que suprime aún más la producción de andrógenos o bloquea sus acciones en la propia célula cancerosa. Otras opciones son la quimioterapia, la radioterapia y los bisfosfonatos, que degradan los osteoclastos y previenen la degradación de los huesos. Los bisfosfonatos todavía están bajo investigación por su efectividad en el cáncer de próstata metastásico. En los casos en los que el paciente tiene un dolor intenso, se puede justificar la administración de narcóticos y la derivación a un especialista en manejo del dolor. Se recomienda que los pacientes con metástasis óseas comiencen la terapia de abstinencia de andrógenos inmediatamente después del diagnóstico. Sin embargo, la terapia hormonal no cura la mayoría de los casos de cáncer de próstata que han hecho metástasis en los huesos. También hay muchos efectos secundarios de la ablación de andrógenos. Es decir, disminución de las funciones sexuales, sofocos, anemia, aumento de peso y, a largo plazo, pérdida de masa ósea y osteoporosis. Un bloqueo total de andrógenos tiene, en el mejor de los casos, beneficios modestos. La tasa de supervivencia se proyecta en un 8-10% a los cinco años y no tiene ninguna ventaja sobre la orquiectomía a los dos años. Además, el 60% de los pacientes recién diagnosticados con metástasis mueren a los 2 años. Se deben realizar exámenes periódicos de rutina y gammagrafías óseas en un paciente con cáncer de próstata metastásico. El objetivo es prevenir la progresión de una metástasis a una fractura patológica. Las metástasis esqueléticas son muy comunes en el cáncer de próstata. En la autopsia, el 84% de las personas con adenocarcinoma de próstata tienen metástasis esqueléticas, mientras que en 1982, de 20.000 nuevos casos de cáncer de próstata, el 21,5% de los pacientes presentaban cáncer de próstata en estadio clínico D (metastásico). Las metástasis esqueléticas también se asocian generalmente con un mal pronóstico. Solo el 23% de los pacientes sobrevivieron 5 años desde el diagnóstico inicial y la tasa de supervivencia a 10 años es del 10%. Como se mencionó anteriormente, los sitios más comunes de metástasis esqueléticas son, en orden de frecuencia decreciente, vértebras, esternón, huesos pélvicos, costillas y fémures1. Sin embargo, los sitios más comunes de fractura son la corteza medial del fémur proximal y el fémur proximal. cuerpos vertebrales porque estos dos sitios deben soportar cargas pesadas. En general, la tasa de fracturas patológicas por cáncer de próstata es relativamente baja en comparación con la de otros cánceres metastásicos.9 La razón de esta tasa baja se debe a la reacción osteoblástica formadora de hueso por parte del tumor. Incluso en aquellos casos en los que se producen fracturas, la tasa de curación se acerca a la del hueso normal. La curación normal, junto con la radioterapia eficaz y la manipulación hormonal, limita la necesidad de estabilización quirúrgica a solo alrededor de una cuarta parte de los pacientes que desarrollan una fractura patológica. Por otro lado, las metástasis esqueléticas que son predominantemente osteolíticas rara vez se observan en el carcinoma de próstata.9 Los autores consideran que las lesiones osteolíticas que se observan en el cáncer de próstata tienen un mayor riesgo de fractura que las metástasis osteoblásticas. Ese riesgo probablemente se acerca al de otros cánceres metastásicos osteolíticos, como el cáncer de mama, el cáncer de pulmón y el cáncer de riñón. Un paciente con una fractura patológica inminente puede tratarse con cirugía, radioterapia, quimioterapia o manipulación hormonal. El tratamiento de una fractura patológica implica tratar la neoplasia al mismo tiempo que se restaura la estabilidad estructural; sin embargo, el tratamiento de uno puede afectar el tratamiento del otro. Por ejemplo, la radioterapia y la quimioterapia pueden tener efectos adversos sobre la cicatrización del hueso. Por esta razón, cuando es necesaria la radioterapia posoperatoria, los reemplazos endoprotésicos en el fémur son el método preferido de estabilización quirúrgica sobre los dispositivos osteosintéticos, ya que no requieren la curación de la fractura. Después de la reconstrucción quirúrgica, se debe permitir que las heridas cicatricen al menos dos semanas antes de comenzar la radioterapia o la quimioterapia para reducir el riesgo de infección. El oncólogo y el cirujano ortopédico deben compartir conjuntamente el seguimiento posoperatorio, ya que alrededor del 25% de estos pacientes desarrollarán otra metástasis ósea que requerirá estabilización quirúrgica. En un estudio realizado por Wedin et al, 26 de 228 (11%) procedimientos para lesiones metastásicas de huesos largos llevaron a fallas que requirieron reoperación. La tasa global de fracaso del cáncer de próstata metastásico solo fue del 10%. De los 26 fracasos, cuatro fueron atribuibles a fracasos inmediatos, seis a progresión tumoral, diez a pseudoartrosis, cinco a fracturas óseas por sobrecarga y uno a luxación tardía de una megaprótesis humeral y necrosis cutánea. Entre 54 endoprótesis, solo un paciente (2%) tuvo una reintervención, mientras que entre 162 operaciones con dispositivos osteosintéticos, 22 (14%) fueron fallas. La tasa de reintervención en los sitios de fractura radiada fue del 13% frente al 10% en los sitios no irradiados. Cinco de los seis pacientes que se sometieron a reoperaciones por progresión del tumor local no habían recibido radioterapia en el sitio de la fractura, y ocho de diez seudoartrosis se habían tratado con radioterapia. Además, los cinco pacientes que tuvieron una fractura por estrés habían sido tratados con radioterapia.9 Todo esto confirma que las endoprótesis tienen una tasa de falla menor que los dispositivos osteosintéticos y que la radiación puede prevenir la progresión del tumor. Tasas de supervivencia del cáncer de próstata
Las tasas de supervivencia proporcionan una idea del porcentaje de personas con el mismo tipo y etapa de cáncer que siguen vivas durante cierto tiempo (generalmente 5 años) después del diagnóstico. Estas tasas no pueden indicar cuánto tiempo usted vivirá, pero pueden ayudarle a tener un mejor entendimiento de cuán probable es que su tratamiento sea eficaz. Tenga en cuenta que las tasas de supervivencia son cálculos que a menudo se basan en los resultados previos de un gran número de personas que padecieron un cáncer específico; sin embargo, no pueden predecir lo que sucederá en el caso particular de una persona. Estas estadísticas pueden ser confusas y pueden ocasionar que tenga más preguntas. Hable con su médico para saber cómo puede que estas estadísticas se apliquen a usted, ya que él o ella está familiarizado con su situación. ¿Qué es tasa relativa de supervivencia a 5 años?Una tasa relativa de supervivencia compara a los hombres que tienen el mismo tipo y etapa de cáncer de próstata con los hombres en la población general. Por ejemplo, si la tasa relativa de supervivencia a 5 años para una etapa específica de cáncer de próstata es 90%, esto significa que los hombres que padecen ese cáncer tienen, en promedio, alrededor de 90% de probabilidades, en comparación con los hombres que no padecen ese cáncer, de vivir al menos 5 años después de recibir el diagnóstico. ¿De dónde provienen estos porcentajes?La Sociedad Americana Contra El Cáncer obtiene la información de la base de datos de SEER (Programa de Vigilancia, Epidemiología y Resultados Finales), mantenida por el Instituto Nacional del Cáncer (NCI), para proporcionar estadísticas de supervivencia para diferentes tipos de cáncer. La base de datos de SEER lleva un registro de las tasas relativas de supervivencia a 5 años para el cáncer de próstata en los Estados Unidos, basándose en cuán lejos se ha propagado el cáncer. Sin embargo, la base de datos de SEER no agrupa a los cánceres según el sistema de estadificación TNM del AJCC (etapa 1, etapa 2, etapa 3, etc.). En cambio, divide a los grupos de cánceres en etapas localizadas, regionales y distantes.
Tasas relativas de supervivencia a 5 años para el cáncer de próstataEstos porcentajes se basan en hombres diagnosticados con cáncer de próstata entre 2010 y 2016.
Cómo entender los porcentajes
|
||||||||||
| Cáncer de pulmón metastásico | El dolor es el síntoma de presentación más común. La fractura patológica rara vez ocurre sin antecedentes de unas pocas semanas o meses de dolor cada vez más intenso. En algunos casos, el paciente ha intentado ignorar o negar los síntomas. A veces, se piensa que una lesión ósea dolorosa es un “tirón muscular” o un “esguince” y se prescriben analgésicos fuertes, lo que permite que el paciente continúe tolerando un dolor muy intenso antes de que se descubra la verdadera naturaleza del problema. También pueden presentarse síntomas sistémicos, como hipercalcemia y osteoartropatía pulmonar hipertrófica (engrosamiento doloroso de los huesos tubulares largos y cortos y dedos en palillo de tambor). | ||||||||||
| Cáncer de riñón metastásico | El dolor es el síntoma de presentación más común. La fractura patológica rara vez ocurre sin antecedentes de unas pocas semanas o meses de dolor cada vez más intenso. En algunos casos, el paciente ha intentado ignorar o negar los síntomas. A veces, se piensa que una lesión ósea dolorosa es un “tirón muscular” o un “esguince” y se prescriben analgésicos fuertes, lo que permite que el paciente continúe tolerando un dolor muy intenso antes de que se descubra la verdadera naturaleza del problema. También pueden aparecer síntomas sistémicos, como hipercalcemia. Un paciente ocasional puede tener hipertesión por el tumor que afecta la vía renina-angiotensina. La hematuria también es un signo común, | ||||||||||
| Condroblastoma | Los pacientes se quejan de dolor punzante en la articulación. Hay mala respuesta a la medicación AINE. Eventualmente aparece una hinchazón o masa. | ||||||||||
| Condroblastoma: pie y tobillo | Los pacientes se quejan de dolor e hinchazón o una masa cerca de la articulación. El dolor puede ser intenso. La articulación cercana puede estar inflamada localmente. | ||||||||||
| Condroblastoma | Los pacientes se quejan de dolor punzante en la articulación. Hay mala respuesta a la medicación AINE. Eventualmente aparece una hinchazón o masa. | ||||||||||
| Fibroma condromixoide | La presentación clínica suele ser dolor crónico, hinchazón y posiblemente una masa de tejido blando palpable o restricción del movimiento. Solo el 5% de los pacientes con CMF presentan una fractura patológica. | ||||||||||
| Fibroma condromixoide: pie y tobillo | Los pacientes se presentan con dolor y una masa de crecimiento lento. | ||||||||||
| Condrosarcoma | La presentación del condrosarcoma depende del grado del tumor. Un tumor de alto grado y de rápido crecimiento puede presentarse con un dolor insoportable. Es más probable que un tumor de grado bajo y más indolente se presente como un paciente mayor que se queja de dolor e hinchazón de la cadera. Los tumores pélvicos se presentan con frecuencia u obstrucción urinaria o pueden enmascararse como “tirones de los músculos de la ingle”. | ||||||||||
| Condrosarcoma – Pie y tobillo | La lesión se presenta como una masa de crecimiento lento con dolor leve. | ||||||||||
| Cordoma | La presentación clínica depende de la ubicación del tumor. Los tumores sacrococcígeos a menudo se presentan como lumbalgia sin patrón característico ni evolución en el tiempo. Los tumores sacrococcígeos también pueden presentarse como disfunción del intestino y la vejiga. Los tumores presacrales a menudo se pueden palpar en el examen rectal, pero esta técnica de examen simple a menudo se pasa por alto. Los tumores del sacro suelen ser grandes en la presentación, ya que un gran volumen de tumor puede acomodarse dentro de la pelvis. Los tumores cervicales anteriores pueden presentarse como disfagia y los tumores cervicales posteriores pueden causar déficits neurológicos. Los tumores en la base del cráneo pueden presentarse con dolores de cabeza. | ||||||||||
| Osteomielitis multifocal recurrente crónica (CRMO) | Los niños se presentan con dolor, dolor profundo, cojera y también pueden presentar fiebre. El área metafisaria de los huesos largos, la clavícula y la cintura escapular son ubicaciones comunes. Se han informado otros sitios como la columna, el tobillo y el pie.
Pueden ocurrir manifestaciones dermatológicas que incluyen psoriasis, acné y pústulas en las palmas de las manos y las plantas de los pies. También se han descrito uveítis y enfermedad inflamatoria intestinal. El síndrome de Majeed consiste en CRMO y aneama diseritropoyético congénito, se ha informado en familias. El gen LPIN2 parece desempeñar un papel en estos casos. |
||||||||||
| Condrosarcoma de células claras | El tumor es una lesión de crecimiento lento que tiene predilección por la epífisis de los huesos largos principales. Por lo tanto, la aparición gradual de dolor relacionado con las articulaciones en la cadera o el hombro es una presentación típica. A veces, una fractura patológica obligará al paciente a ver tratamiento para la lesión, a pesar de que ha habido un dolor leve o moderado durante años. La presentación general de esta lesión imita la de un tumor óseo benigno. | ||||||||||
| Fibroma colágeno (fibroblastoma desmoplásico) | El paciente presenta una masa indolora de crecimiento lento en los tejidos subcuticulares, un sitio periférico, más comúnmente en las extremidades superiores o inferiores, que pueden afectar al músculo esquelético. El tamaño puede ser de uno a varios centímetros, con masas tan grandes como sea posible de 20 cm. | ||||||||||
| Condroblastoma: pie y tobillo | Los pacientes se quejan de dolor e hinchazón o una masa cerca de la articulación. El dolor puede ser intenso. La articulación cercana puede estar inflamada localmente. | ||||||||||
| Condroma perióstico | Los síntomas están presentes durante 1 a 5 años. Los pacientes se quejan de una masa o hinchazón dolorosa. | ||||||||||
| Condroma perióstico – pie y tobillo | La lesión se presenta con dolor e hinchazón. |
| Condromatosis sinovial | La condromatosis sinovial se presenta como la aparición gradual de dolor y rigidez monoarticular. Si se permite que continúen, los síntomas lentos y progresivos pueden resultar en una disminución del rango de movimiento, derrames, crepitaciones y eventual bloqueo de la articulación. Esta condición también se ha descrito en la membrana sinovial, tejidos blandos y bursas. La condromatosis sinovial secundaria puede estar presente después de una osteoartritis de larga duración. Existen varias etiologías posibles de la condromatosis sinovial. El trauma está respaldado por el hecho de que el proceso ocurre principalmente en las articulaciones dominantes de las extremidades superiores y en las que soportan peso. La infección es otro posible culpable. Otra teoría postula que el cartílago desprendido hacia la articulación es absorbido por la membrana sinovial. |
| Condrosarcoma | La presentación del condrosarcoma depende del grado del tumor. Un tumor de alto grado y de rápido crecimiento puede presentarse con un dolor insoportable. Es más probable que un tumor de grado bajo y más indolente se presente como un paciente mayor que se queja de dolor e hinchazón de la cadera. Los tumores pélvicos se presentan con frecuencia u obstrucción urinaria o pueden enmascararse como “tirones de los músculos de la ingle”. |
| Condrosarcoma – Pie y tobillo | La lesión se presenta como una masa de crecimiento lento con dolor leve. |
| Condrosarcoma mixoide extraesquelético | Aproximadamente el 80% de estos tumores se presentan en las extremidades y el 20% en el tronco. La extremidad inferior es el sitio más común. Los pacientes se presentan con hinchazón y el tumor puede ser bastante voluminoso. |
| Condrosarcoma secundario | |
| Cordoma | La presentación clínica depende de la ubicación del tumor. Los tumores sacrococcígeos a menudo se presentan como lumbalgia sin patrón característico ni evolución en el tiempo. Los tumores sacrococcígeos también pueden presentarse como disfunción del intestino y la vejiga. Los tumores presacrales a menudo se pueden palpar en el examen rectal, pero esta técnica de examen simple a menudo se pasa por alto. Los tumores del sacro suelen ser grandes en la presentación, ya que un gran volumen de tumor puede acomodarse dentro de la pelvis. Los tumores cervicales anteriores pueden presentarse como disfagia y los tumores cervicales posteriores pueden causar déficits neurológicos. Los tumores en la base del cráneo pueden presentarse con dolores de cabeza. |
| Condrosarcoma desdiferenciado | Los pacientes suelen presentar dolor y una masa. Aproximadamente el 90% de los pacientes presentan dolor. La naturaleza agresiva del sarcoma se refleja típicamente en un crecimiento rápido, una fractura patológica o un dolor intenso y creciente. Aproximadamente el 12% de los pacientes tienen fractura patológica. |
| Liposarcoma desdiferenciado | Los pacientes se presentan con una masa agrandada. Puede ser suave y carnoso y relativamente indoloro. Algunos casos pueden volverse muy grandes y permanecer indoloros. |
| Fibroma desmoplásico | Los hallazgos clínicos incluyen dolor al final del curso clínico e hinchazón. Puede presentarse como un derrame si está cerca de una articulación. Solo el 12% presenta una fractura patológica. |
| Estenosis medular diafisaria con histiocitoma fibroso maligno | |
| Estenosis medular diafisaria con histiocitoma fibroso maligno | |
| Displasia fibrosa | La displasia fibrosa monostótica puede ser completamente asintomática y, a menudo, es un hallazgo incidental en la radiografía. También puede haber dolor e hinchazón en el sitio de la lesión. Las pacientes femeninas pueden tener un aumento de los síntomas durante el embarazo. Desafortunadamente, este tumor también puede presentarse como una fractura patológica seguida de una pseudoartrosis o consolidación defectuosa. |
| Displasia osteofibrosa | El tumor se presenta clínicamente como una masa que aumenta de tamaño e indolora. |
| Anomalía del drenaje venoso intraóseo | |
| Fibroma condromixoide: pie y tobillo | Los pacientes se presentan con dolor y una masa de crecimiento lento. |
| Granuloma anular – pie y tobillo | GA localizada y diseminada son los dos tipos más comunes.
La AG localizada comprende aproximadamente 3 de cada 4 casos. Se observan una o varias pápulas pequeñas, firmes, de color carne o eyrhematosas, que pueden formar un anillo y expandirse lentamente en diámetro. Estas lesiones ocurren típicamente en las superficies lateral y dorsal de las manos y los pies. La GA diseminada o generalizada se caracteriza por lesiones diseminadas de naturaleza similar a las de la GA localizada. Las lesiones pueden durar varios años. Las lesiones pueden mejorar en invierno y empeorar en verano. La GA subcutánea es más común en niños de 2 a 5 años. Esta forma se manifiesta con mayor frecuencia como una gran masa de tejido blando asintomática. Aunque los nódulos suelen ser estables durante meses, pueden agrandarse rápidamente en el transcurso de semanas. Los pacientes con AG subcutánea se presentan con un nódulo firme, no doloroso, de color carne o rosado sin alteración epidérmica suprayacente. Las lesiones son típicamente solitarias pero pueden ocurrir en grupos. El sitio de afectación más comúnmente informado son las extremidades inferiores (65% de los casos), a menudo en la superficie pretibial. Otros sitios típicos incluyen los dedos y las palmas y el dorso de los pies. Las nalgas, la frente y el cuero cabelludo se ven afectados con menos frecuencia. Los nódulos dérmicos o subcutáneos profundos en las extremidades están adheridos a la fascia y, por lo tanto, a menudo son móviles. mientras que las lesiones en el cuero cabelludo están adheridas al periostio subyacente y, por lo tanto, son fijas o solo ligeramente móviles. Estas lesiones no progresan a una enfermedad sistémica. La perforación es un tipo raro, más común en las mujeres y se encuentra en las extremidades superiores y la pelvis, el abdomen, el tronco y las extremidades. Las lesiones pueden causar picazón y dolor. |
| Melanoma maligno: pie y tobillo | |
| Sarcoma de Ewing – Pie y tobillo | Los pacientes normalmente tienen dolor acompañado de una hinchazón difusa significativa. El área puede aparecer inflamada y se puede hacer un diagnóstico erróneo de infección. El recuento de glóbulos blancos, la velocidad de sedimentación globular y la temperatura pueden estar moderadamente elevados.
Los pacientes presentan síntomas durante un promedio de 14 meses antes del diagnóstico. Los pacientes con lesiones en el retropié tienen una duración promedio de los síntomas de 22 meses antes del diagnóstico, mientras que aquellos con lesiones en el antepié tienen una duración promedio de los síntomas de siete meses. |
| Sarcoma epitelioide: pie y tobillo | El tumor puede presentarse como un nódulo pequeño, firme superficial o profundo o un grupo focal de nódulos. La presentación multifocal regional es una característica inusual que presenta este tumor. Con frecuencia, este tumor se diagnostica erróneamente como una afección de la piel, verrugas o callos, y un diagnóstico correcto puede retrasarse con graves consecuencias médicas y legales. Aproximadamente la mitad de los tumores no son dolorosos. El tumor se presenta tanto en el tejido subcutáneo como en los tejidos más profundos. Cuando se localiza en el subcutis, generalmente se presenta como un nódulo firme que puede ser solitario o múltiple, tiene una consistencia similar a un callo y a menudo se describe como un “nudo duro leñoso” o: bulto firme ”de crecimiento lento e indoloro. Los nódulos situados en la dermis a menudo se elevan por encima de la superficie de la piel y con frecuencia se ulceran semanas o meses después de que se notan por primera vez. Estas lesiones a menudo se diagnostican erróneamente como una “úlcera indurada”, un “absceso que drena” o una “verruga infectada” que no cicatriza a pesar de la terapia intensiva. La mayoría de los tumores tienen de 3 a 6 cm de diámetro. |
| Sarcoma sinovial: pie y tobillo | La presentación del sarcoma sinovial es variable y puede simular un proceso benigno como el quiste ganglionar. El paciente puede tener una masa que ha estado presente durante meses, años o incluso décadas, con crecimiento lento y poco o ningún síntoma. Es posible que haya habido un crecimiento rápido reciente de una lesión que ha estado presente durante años sin cambios aparentes. Por el contrario, algunos de estos sarcomas pueden ser muy dolorosos desde el principio. La duración media de los síntomas antes del diagnóstico es de 21 meses. |
| Síndrome de Ollier | El niño tiene un crecimiento anormal, acortamiento y deformidad de la extremidad afectada. |
| Encondroma | La mayoría de los pacientes no presentan síntomas. La presentación más común es un paciente que se ha lesionado la rodilla o el hombro y al que se le realiza una radiografía, lo que lleva al descubrimiento de la lesión previamente asintomática. Las lesiones en las manos y los pies pueden debilitar el hueso y causar dolor, hinchazón y pequeñas fracturas patológicas durante las actividades. |
| Encondroma – Pie y tobillo | Los pacientes presentan dolor durante las actividades o después de una lesión, pero rara vez se palpa una masa en la exploración física. Para encondromas en las falanges, la fractura patológica a través de la lesión hará que el paciente busque atención médica. Algunos pacientes tienen múltiples encondromas y requieren atención especial y posiblemente derivación a un especialista en tumores óseos. |
| Encondroma | La mayoría de los pacientes no presentan síntomas. La presentación más común es un paciente que se ha lesionado la rodilla o el hombro y al que se le realiza una radiografía, lo que lleva al descubrimiento de la lesión previamente asintomática. Las lesiones en las manos y los pies pueden debilitar el hueso y causar dolor, hinchazón y pequeñas fracturas patológicas durante las actividades. |
| Encondroma – Pie y tobillo | Los pacientes presentan dolor durante las actividades o después de una lesión, pero rara vez se palpa una masa en la exploración física. Para encondromas en las falanges, la fractura patológica a través de la lesión hará que el paciente busque atención médica. Algunos pacientes tienen múltiples encondromas y requieren atención especial y posiblemente derivación a un especialista en tumores óseos. |
| Enfermedad de Paget | El sarcoma de Paget generalmente se presenta como un dolor nuevo y progresivo en un paciente con enfermedad de Paget de larga duración. Otros síntomas pueden incluir hinchazón de tejidos blandos o fracturas patológicas. La fosfatasa alcalina sérica que se eleva con la enfermedad de Paget puede aumentar aún más con la aparición del sarcoma. |
| Granuloma eosinofílico | EG es normalmente sintomático. El dolor local, la hinchazón y la sensibilidad son comunes y la VSG puede estar elevada. |
| Hemangioendotelioma epitelioide | Clínicamente, los hemangioendoteliomas epitelioides se presentan con dolor e hinchazón. Si está presente en la columna, una lesión puede causar síntomas radiculares o paraplejía. |
| Hemangioendotelioma epitelioide | Clínicamente, los hemangioendoteliomas epitelioides se presentan con dolor e hinchazón. Si está presente en la columna, una lesión puede causar síntomas radiculares o paraplejía. |
| Hemangioma epitelioide | Clínicamente, los pacientes suelen presentar dolor en los sitios afectados. |
| Sarcoma epitelioide | El tumor puede presentarse como un nódulo pequeño, firme superficial o profundo o un grupo focal de nódulos. La presentación multifocal regional es una característica inusual que presenta este tumor. Con frecuencia, este tumor se diagnostica erróneamente como una afección de la piel, verrugas o callos, y un diagnóstico correcto puede retrasarse con graves consecuencias médicas y legales. Aproximadamente la mitad de los tumores no son dolorosos. El tumor se presenta tanto en el tejido subcutáneo como en los tejidos más profundos. Cuando se localiza en el subcutis, generalmente se presenta como un nódulo firme que puede ser solitario o múltiple, tiene una consistencia similar a un callo y a menudo se describe como un “nudo duro leñoso” o: bulto firme ”de crecimiento lento e indoloro. Los nódulos situados en la dermis a menudo se elevan por encima de la superficie de la piel y con frecuencia se ulceran semanas o meses después de que se notan por primera vez. Estas lesiones a menudo se diagnostican erróneamente como una “úlcera indurada”, un “absceso que drena” o una “verruga infectada” que no cicatriza a pesar de la terapia intensiva. La mayoría de los tumores tienen de 3 a 6 cm de diámetro.
La metástasis en los ganglios linfáticos debe evaluarse en el momento de la presentación, debido a la inusual propensión de este tumor a diseminarse a los ganglios regionales. En general, las metástasis en los ganglios linfáticos en los sarcomas son bastante raras y ocurren en aproximadamente el 2,6% de todos los pacientes con sarcoma en series grandes. En ES, sin embargo, la tasa de metástasis en los ganglios linfáticos es de alrededor del 30% (rango 14-44%). La metástasis pulmonar ocurre en el 20-40% de los pacientes. |
| Sarcoma epitelioide: pie y tobillo | El tumor puede presentarse como un nódulo pequeño, firme superficial o profundo o un grupo focal de nódulos. La presentación multifocal regional es una característica inusual que presenta este tumor. Con frecuencia, este tumor se diagnostica erróneamente como una afección de la piel, verrugas o callos, y un diagnóstico correcto puede retrasarse con graves consecuencias médicas y legales. Aproximadamente la mitad de los tumores no son dolorosos. El tumor se presenta tanto en el tejido subcutáneo como en los tejidos más profundos. Cuando se localiza en el subcutis, generalmente se presenta como un nódulo firme que puede ser solitario o múltiple, tiene una consistencia similar a un callo y a menudo se describe como un “nudo duro leñoso” o: bulto firme ”de crecimiento lento e indoloro. Los nódulos situados en la dermis a menudo se elevan por encima de la superficie de la piel y con frecuencia se ulceran semanas o meses después de que se notan por primera vez. Estas lesiones a menudo se diagnostican erróneamente como una “úlcera indurada”, un “absceso que drena” o una “verruga infectada” que no cicatriza a pesar de la terapia intensiva. La mayoría de los tumores tienen de 3 a 6 cm de diámetro. |
| Enfermedad de Erdheim-Chester (ECD) | La mayoría de los pacientes se presentan con dolor en las extremidades inferiores. La afectación suele ser bilateral y simétrica. Existe una afectación específica del esqueleto apendicular, aunque también se ha informado la afectación del esqueleto axial. Puede haber síntomas generales asociados como fiebre, mialgia, pérdida de peso, sudores nocturnos y síntomas similares a los de la gripe. El paciente con afectación sistémica puede presentar síntomas neurológicos, diabetes insípida, exoftalmos, xantomas cutáneos y lumbalgia por fibrosis retroperitoneal. |
| Sarcoma de Ewing – Pie y tobillo | Los pacientes normalmente tienen dolor acompañado de una hinchazón difusa significativa. El área puede aparecer inflamada y se puede hacer un diagnóstico erróneo de infección. El recuento de glóbulos blancos, la velocidad de sedimentación globular y la temperatura pueden estar moderadamente elevados.
Los pacientes presentan síntomas durante un promedio de 14 meses antes del diagnóstico. Los pacientes con lesiones en el retropié tienen una duración promedio de los síntomas de 22 meses antes del diagnóstico, mientras que aquellos con lesiones en el antepié tienen una duración promedio de los síntomas de siete meses. |
| Sarcoma de Ewing | La presentación clínica del sarcoma de Ewing incluye dolor e hinchazón de semanas o meses de duración. A veces se observan eritema y calor en el área local. La osteomielitis es a menudo el diagnóstico inicial basado en fiebres intermitentes, leucocitosis, anemia y aumento de la VSG. |
| Osteosarcoma extraesquelético | |
| Condrosarcoma mixoide extraesquelético | Aproximadamente el 80% de estos tumores se presentan en las extremidades y el 20% en el tronco. La extremidad inferior es el sitio más común. Los pacientes se presentan con hinchazón y el tumor puede ser bastante voluminoso. |
| Osteosarcoma extraesquelético | |
| Fibroma condromixoide | La presentación clínica suele ser dolor crónico, hinchazón y posiblemente una masa de tejido blando palpable o restricción del movimiento. Solo el 5% de los pacientes con CMF presentan una fractura patológica. |
| Fibroma desmoplásico | Los hallazgos clínicos incluyen dolor al final del curso clínico e hinchazón. Puede presentarse como un derrame si está cerca de una articulación. Solo el 12% presenta una fractura patológica. |
| Fibroma plantar – pie y tobillo | La mayoría de los pacientes son asintomáticos, pero algunos tienen dolor relacionado con la actividad. Entre un tercio y la mitad de los pacientes tienen nódulos bilaterales. El dolor se presenta con las actividades que soportan peso. Cuando las lesiones son lo suficientemente grandes como para presionar los nervios plantares, puede haber entumecimiento o disestesia en las porciones distales del pie. En un paciente pediátrico, una gran lesión provocó la contractura de los tendones flexores del dedo gordo y la pérdida de la extensión de los dedos inferiores. |
| Fibrosarcoma | La presentación clínica más común es la de una masa dolorosa localizada. |
| Displasia fibrosa | La displasia fibrosa monostótica puede ser completamente asintomática y, a menudo, es un hallazgo incidental en la radiografía. También puede haber dolor e hinchazón en el sitio de la lesión. Las pacientes femeninas pueden tener un aumento de los síntomas durante el embarazo. Desafortunadamente, este tumor también puede presentarse como una fractura patológica seguida de una pseudoartrosis o consolidación defectuosa. |
| Periostitis florida reactiva | El examen revela una masa sensible con leve calor circundante y eritema, pero sin fiebre ni adenopatía. Los estudios de laboratorio suelen mostrar un recuento y diferencial de glóbulos blancos normales y una tasa de sedementación levemente elevada. Los electrolitos y otros valores de laboratorio no se ven afectados. Las radiografías simples muestran que el tumor está adyacente al hueso en lugar de surgir de él. Se observa inflamación de los tejidos blandos y una marcada reacción perióstica. Puede haber formación de hueso nuevo en los tejidos blandos y adelgazamiento cortical sutil. En casos raros hay erosión local de la corteza. Las gammagrafías óseas con tecnecio-99 muestran un foco solitario de captación intensa. Las exploraciones por resonancia magnética muestran una masa y anomalías de señal llamativas en los tejidos blandos, así como en la médula ósea cercana, que son relativamente inespecíficas y son compatibles con una infección, un traumatismo o un tumor. |
| Quiste de ganglio – pie y tobillo | La mayoría de los quistes ganglionares pueden diagnosticarse definitivamente basándose en una historia clínica y un examen físico cuidadosos. Según la historia, la lesión tiende tanto a aumentar como a disminuir de tamaño con el tiempo. En el examen, el quiste de ganglio generalmente tiene una ubicación superficial y puede estar adyacente a una articulación. Cuando estas lesiones ocurren cerca de una articulación, a menudo se observa osteoartritis en la radiografía. La masa es blanda cuando la articulación cercana está relajada y se vuelve firme cuando la articulación o el músculo cercanos se tensan. La luz de un bolígrafo o un pequeño puntero láser transiluminarán el quiste. |
| Quiste de ganglio – pie y tobillo | La mayoría de los quistes ganglionares pueden diagnosticarse definitivamente basándose en una historia clínica y un examen físico cuidadosos. Según la historia, la lesión tiende tanto a aumentar como a disminuir de tamaño con el tiempo. En el examen, el quiste de ganglio generalmente tiene una ubicación superficial y puede estar adyacente a una articulación. Cuando estas lesiones ocurren cerca de una articulación, a menudo se observa osteoartritis en la radiografía. La masa es blanda cuando la articulación cercana está relajada y se vuelve firme cuando la articulación o el músculo cercanos se tensan. La luz de un bolígrafo o un pequeño puntero láser transiluminarán el quiste. |
| Granuloma reparador de células gigantes | Dolor e hinchazón o una leve fractura patológica. |
| Tumor de células gigantes | La mayoría de los pacientes se presentan con dolor de progresión lenta, con o sin una masa. Los síntomas surgen cuando la lesión comienza a destruir la corteza e irrita el periostio o cuando el debilitamiento del hueso causado por el tumor provoca dolor por fractura patológica inminente. Algunos tumores de células gigantes se presentan con una fractura patológica. |
| Tumor de células gigantes: pie y tobillo | El paciente típico tiene antecedentes de dolor que aumenta gradualmente. En el pie puede aparecer una masa debido a la limitación de los tejidos blandos. El dolor por fractura patológica o microfractura puede hacer que el paciente busque tratamiento. |
| Tumor de células gigantes de la vaina del tendón | |
| Tumor de células gigantes de la vaina del tendón: pie y tobillo | Clínicamente, los pacientes refieren una masa solitaria firme, indolora y de crecimiento lento adyacente a los tendones dorsales o plantares, las articulaciones del mediopié o la articulación del tobillo, que ha estado presente durante uno o dos años en promedio. Puede haber antecedentes de trauma y los síntomas neurológicos ocurren raramente. En un estudio, las lesiones en el antepié ocurrieron en el primer, segundo y quinto rayos exclusivamente, lo que indica que puede haber alguna relación entre la carga de peso y este tumor. El tumor puede causar o acentuar una deformidad angular como hallux valgus. |
| Condromatosis sinovial gigante | En la serie del autor, el tumor se ha encontrado como una lesión incidental en el tobillo, una masa dolorosa en el tobillo o la rodilla o como una sensación de chasquido en la cadera. Se han informado lesiones en la mayoría de las articulaciones grandes, incluidos el codo, el hombro y la extremidad inferior. Algunos tumores de la serie del autor se han localizado en una vaina tendinosa fuera de la articulación por completo, pero anatómicamente no muy lejos de una articulación grande. |
| Condromatosis sinovial gigante | En la serie del autor, el tumor se ha encontrado como una lesión incidental en el tobillo, una masa dolorosa en el tobillo o la rodilla o como una sensación de chasquido en la cadera. Se han informado lesiones en la mayoría de las articulaciones grandes, incluidos el codo, el hombro y la extremidad inferior. Algunos tumores de la serie del autor se han localizado en una vaina tendinosa fuera de la articulación por completo, pero anatómicamente no muy lejos de una articulación grande. |
| Tumor de células gigantes de la vaina del tendón: pie y tobillo | Clínicamente, los pacientes refieren una masa solitaria firme, indolora y de crecimiento lento adyacente a los tendones dorsales o plantares, las articulaciones del mediopié o la articulación del tobillo, que ha estado presente durante uno o dos años en promedio. Puede haber antecedentes de trauma y los síntomas neurológicos ocurren raramente. En un estudio, las lesiones en el antepié ocurrieron en el primer, segundo y quinto rayos exclusivamente, lo que indica que puede haber alguna relación entre la carga de peso y este tumor. El tumor puede causar o acentuar una deformidad angular como hallux valgus. |
| Tumor glómico | Clínicamente, los tumores glómicos se caracterizan por una tríada de sensibilidad al frío, sensibilidad localizada y dolor intermitente intenso. El dolor puede ser insoportable y se describe como ardor o estallido. La causa exacta del dolor no se comprende completamente, pero se han identificado fibras nerviosas que contienen la sustancia neurotransmisora del dolor P en el tumor. |
| Tumor glómico – tejido blando | En la presentación inicial, la lesión puede no ser palpable y la decoloración azul-púrpura característica puede no ser visible. El paciente informa de un área dolorosa que puede estar muy bien localizada.
Clínicamente, los tumores glómicos se caracterizan por una tríada de sensibilidad al frío, sensibilidad localizada y dolor intermitente intenso. El dolor puede ser insoportable y se describe como ardor o estallido. La causa exacta del dolor no se comprende completamente, pero se han identificado fibras nerviosas que contienen la sustancia neurotransmisora del dolor P en el tumor. Una prueba de diagnóstico es la sensibilidad a la presión con un objeto muy pequeño, como la cabeza de un alfiler o un alambre de Kirschner sin filo fino. La sensibilidad al frío se puede probar con baños de hielo. Se dice que la aplicación de un torniquete alivia el dolor, que vuelve rápidamente cuando se suelta el torniquete. La lesión puede tener un color azul visible. |
| Sarcoma granulocítico en hueso | La mayoría de los sarcomas granulocíticos óseos se presentan con dolor esquelético atribuible a la ubicación de la lesión. Este tumor tiene tres presentaciones clínicas características: una, en un individuo sin enfermedad conocida donde es un presagio de leucemia mielógena aguda; dos, en un individuo con un trastorno mieloproliferativo ya conocido. La evaluación posterior de la biopsia de sangre periférica y médula ósea en este caso demostró que se trataba de un sarcoma granulocítico sin evidencia sistémica de leucemia mielógena aguda. La aparición del tumor en un individuo previamente sano presenta un importante desafío diagnóstico y el 75% de estos casos suelen ser mal diagnosticados. |
| Granuloma anular – pie y tobillo | GA localizada y diseminada son los dos tipos más comunes.
La AG localizada comprende aproximadamente 3 de cada 4 casos. Se observan una o varias pápulas pequeñas, firmes, de color carne o eyrhematosas, que pueden formar un anillo y expandirse lentamente en diámetro. Estas lesiones ocurren típicamente en las superficies lateral y dorsal de las manos y los pies. La GA diseminada o generalizada se caracteriza por lesiones diseminadas de naturaleza similar a las de la GA localizada. Las lesiones pueden durar varios años. Las lesiones pueden mejorar en invierno y empeorar en verano. Subcutaneous GA is most common in children 2 to 5 years old. This form most often manifests as a large, asymptomatic soft tissue mass. Although nodules are usually stable for months, they may rapidly enlarge over the course of weeks. Patients with subcutaneous GA present with a firm, nontender, flesh-colored or pinkish nodule without overlying epidermal alteration .Lesions are typically solitary but may occur in clusters. The most commonly reported site of involvement is the lower extremities (65% of cases), often on the pretibial surface. Other typical sites include the fingers and palms and the dorsa of the feet. The buttocks, forehead, and scalp are less commonly affected. Deep dermal or subcutaneous nodules on the extremities are attached to fascia and are often therefore mobile, whereas lesions on the scalp are attached to underlying periosteum and are therefore fixed or only slightly mobile. These lesions do not progress to a systemic illness. La perforación es un tipo raro, más común en las mujeres y se encuentra en las extremidades superiores y la pelvis, el abdomen, el tronco y las extremidades. Las lesiones pueden causar picazón y dolor. |
| Tumor de células gigantes de la vaina del tendón | |
| Granuloma eosinofílico | EG es normalmente sintomático. El dolor local, la hinchazón y la sensibilidad son comunes y la VSG puede estar elevada. |
| Condrosarcoma mesenquimatoso | Los pacientes se presentan con dolor local y una masa. El dolor de cabeza ocurre en las lesiones del cráneo. Las lesiones de la base del cráneo se presentan con síntomas del tronco encefálico y disfunción de los pares craneales. |
| Cáncer de mama metastásico | El dolor es el síntoma de presentación más común. La fractura patológica rara vez ocurre sin antecedentes de unas pocas semanas o meses de dolor cada vez más intenso. En algunos casos, el paciente ha intentado ignorar o negar los síntomas. A veces, se piensa que una lesión ósea dolorosa es un “tirón muscular” o un “esguince” y se prescriben analgésicos fuertes, lo que permite que el paciente continúe tolerando un dolor muy intenso antes de que se descubra la verdadera naturaleza del problema. También pueden aparecer síntomas sistémicos, como hipercalcemia. |
| Cáncer de riñón metastásico | El dolor es el síntoma de presentación más común. La fractura patológica rara vez ocurre sin antecedentes de unas pocas semanas o meses de dolor cada vez más intenso. En algunos casos, el paciente ha intentado ignorar o negar los síntomas. A veces, se piensa que una lesión ósea dolorosa es un “tirón muscular” o un “esguince” y se prescriben analgésicos fuertes, lo que permite que el paciente continúe tolerando un dolor muy intenso antes de que se descubra la verdadera naturaleza del problema. También pueden aparecer síntomas sistémicos, como hipercalcemia. Un paciente ocasional puede tener hipertesión por el tumor que afecta la vía renina-angiotensina. La hematuria también es un signo común, |
| Cáncer de pulmón metastásico | El dolor es el síntoma de presentación más común. La fractura patológica rara vez ocurre sin antecedentes de unas pocas semanas o meses de dolor cada vez más intenso. En algunos casos, el paciente ha intentado ignorar o negar los síntomas. A veces, se piensa que una lesión ósea dolorosa es un “tirón muscular” o un “esguince” y se prescriben analgésicos fuertes, lo que permite que el paciente continúe tolerando un dolor muy intenso antes de que se descubra la verdadera naturaleza del problema. También pueden presentarse síntomas sistémicos, como hipercalcemia y osteoartropatía pulmonar hipertrófica (engrosamiento doloroso de los huesos tubulares largos y cortos y dedos en palillo de tambor). |
| Cáncer de próstata metastásico | El diagnóstico de cáncer de próstata generalmente comienza con PSA sérico elevado y / o fosfatasa ácida prostática, o la palpación de nódulos en un tacto rectal. El diagnóstico generalmente se confirma mediante una biopsia guiada por ecografía transrectal. El PSA también es actualmente el marcador más útil para evaluar el nivel de afectación ósea en el cáncer de próstata. El diagnóstico de metástasis óseas se realiza mediante una gammagrafía ósea. El problema con las gammagrafías óseas es que se consideran sensibles en el diagnóstico inicial, pero la especificidad es relativamente baja. Tampoco son adecuados para controlar la respuesta al tratamiento a corto plazo. Actualmente se están investigando otros marcadores para medir la cantidad de afectación ósea. Uno de estos marcadores, el telopéptido carboxi-terminal reticulado de piridinolina de colágeno tipo I (ICTP), se escinde durante la degradación del colágeno tipo I, lo que es indicativo de recambio óseo. El pensamiento actual es que el ICTP es más específico para el hueso que el PSA, pero es necesario realizar más estudios.
Se desconoce el mecanismo real por el cual las células cancerosas metastásicas provocan reacciones osteoblásticas. Sin embargo, Koeneman et al han propuesto que “para prosperar en el entorno óseo, las células cancerosas deben adquirir propiedades ‘similares a las de las células óseas'”. Se cree que existen interacciones celulares recíprocas entre las células del cáncer de próstata y el estroma óseo que dan como resultado la proliferación tanto del cáncer de próstata como de las células del estroma óseo. Esta interacción implica la participación de factores de crecimiento, como TGF-ß, bFGF e IGF tanto en el hueso como en la próstata, que se modula además por las acciones diferenciadoras de las proteínas morfogenéticas óseas y las proteínas relacionadas con la PTH. Al final, las condiciones son adecuadas para el crecimiento osteoblástico y las consecuencias son dolor óseo, inflamación y un mayor riesgo de fracturas. Los tumores vertebrales pueden comprimir la médula espinal y causar daño a los nervios. Es importante tener una buena idea del nivel de afectación ósea, ya que tiene un impacto significativo en la supervivencia general del paciente. Los métodos actuales de tratamiento para el cáncer de próstata son la prostatectomía, la radioterapia y la terapia hormonal. La terapia hormonal se puede lograr mediante algunos métodos diferentes. La orquiectomía y la administración de estrógenos son dos opciones. Aunque el 60-80% de los pacientes con cáncer de próstata avanzado mejoran después de la castración, hay una progresión inevitable a un estado independiente de andrógenos testicular, en el que los andrógenos suprarrenales se hacen cargo. También existen fármacos que afectan directamente a la glándula pituitaria, como los agonistas de LHRH, y algunos pacientes también reciben fármacos antiandrógenos para bloquear el efecto de los andrógenos producidos por las glándulas suprarrenales produciendo un bloqueo total de andrógenos. Algunos pacientes con metástasis a menudo requieren un tratamiento adicional, como la terapia hormonal de segunda línea, que suprime aún más la producción de andrógenos o bloquea sus acciones en la propia célula cancerosa. Otras opciones son la quimioterapia, la radioterapia y los bisfosfonatos, que degradan los osteoclastos y previenen la degradación de los huesos. Los bisfosfonatos todavía están bajo investigación por su efectividad en el cáncer de próstata metastásico. En los casos en los que el paciente tiene un dolor intenso, se puede justificar la administración de narcóticos y la derivación a un especialista en manejo del dolor. Se recomienda que los pacientes con metástasis óseas comiencen la terapia de abstinencia de andrógenos inmediatamente después del diagnóstico. Sin embargo, la terapia hormonal no cura la mayoría de los casos de cáncer de próstata que han hecho metástasis en los huesos. También hay muchos efectos secundarios de la ablación de andrógenos. Es decir, disminución de las funciones sexuales, sofocos, anemia, aumento de peso y, a largo plazo, pérdida de masa ósea y osteoporosis. Un bloqueo total de andrógenos tiene, en el mejor de los casos, beneficios modestos. La tasa de supervivencia se proyecta en un 8-10% a los cinco años y no tiene ninguna ventaja sobre la orquiectomía a los dos años. Además, el 60% de los pacientes recién diagnosticados con metástasis mueren a los 2 años. Se deben realizar exámenes periódicos de rutina y gammagrafías óseas en un paciente con cáncer de próstata metastásico. El objetivo es prevenir la progresión de una metástasis a una fractura patológica. Las metástasis esqueléticas son muy comunes en el cáncer de próstata. En la autopsia, el 84% de las personas con adenocarcinoma de próstata tienen metástasis esqueléticas, mientras que en 1982, de 20.000 nuevos casos de cáncer de próstata, el 21,5% de los pacientes presentaban cáncer de próstata en estadio clínico D (metastásico). Las metástasis esqueléticas también se asocian generalmente con un mal pronóstico. Solo el 23% de los pacientes sobrevivieron 5 años desde el diagnóstico inicial y la tasa de supervivencia a 10 años es del 10%. Como se mencionó anteriormente, los sitios más comunes de metástasis esqueléticas son, en orden de frecuencia decreciente, vértebras, esternón, huesos pélvicos, costillas y fémures1. Sin embargo, los sitios más comunes de fractura son la corteza medial del fémur proximal y el fémur proximal. cuerpos vertebrales porque estos dos sitios deben soportar cargas pesadas. En general, la tasa de fracturas patológicas por cáncer de próstata es relativamente baja en comparación con la de otros cánceres metastásicos.9 La razón de esta tasa baja se debe a la reacción osteoblástica formadora de hueso por parte del tumor. Incluso en aquellos casos en los que se producen fracturas, la tasa de curación se acerca a la del hueso normal. La curación normal, junto con la radioterapia eficaz y la manipulación hormonal, limita la necesidad de estabilización quirúrgica a solo alrededor de una cuarta parte de los pacientes que desarrollan una fractura patológica. Por otro lado, las metástasis esqueléticas que son predominantemente osteolíticas rara vez se observan en el carcinoma de próstata.9 Los autores consideran que las lesiones osteolíticas que se observan en el cáncer de próstata tienen un mayor riesgo de fractura que las metástasis osteoblásticas. Ese riesgo probablemente se acerca al de otros cánceres metastásicos osteolíticos, como el cáncer de mama, el cáncer de pulmón y el cáncer de riñón. Un paciente con una fractura patológica inminente puede tratarse con cirugía, radioterapia, quimioterapia o manipulación hormonal. El tratamiento de una fractura patológica implica tratar la neoplasia al mismo tiempo que se restaura la estabilidad estructural; sin embargo, el tratamiento de uno puede afectar el tratamiento del otro. Por ejemplo, la radioterapia y la quimioterapia pueden tener efectos adversos sobre la cicatrización del hueso. Por esta razón, cuando es necesaria la radioterapia posoperatoria, los reemplazos endoprotésicos en el fémur son el método preferido de estabilización quirúrgica sobre los dispositivos osteosintéticos, ya que no requieren la curación de la fractura. Después de la reconstrucción quirúrgica, se debe permitir que las heridas cicatricen al menos dos semanas antes de comenzar la radioterapia o la quimioterapia para reducir el riesgo de infección. El oncólogo y el cirujano ortopédico deben compartir conjuntamente el seguimiento posoperatorio, ya que alrededor del 25% de estos pacientes desarrollarán otra metástasis ósea que requerirá estabilización quirúrgica. En un estudio realizado por Wedin et al, 26 de 228 (11%) procedimientos para lesiones metastásicas de huesos largos llevaron a fallas que requirieron reoperación. La tasa global de fracaso del cáncer de próstata metastásico solo fue del 10%. De los 26 fracasos, cuatro fueron atribuibles a fracasos inmediatos, seis a progresión tumoral, diez a pseudoartrosis, cinco a fracturas óseas por sobrecarga y uno a luxación tardía de una megaprótesis humeral y necrosis cutánea. Entre 54 endoprótesis, solo un paciente (2%) tuvo una reintervención, mientras que entre 162 operaciones con dispositivos osteosintéticos, 22 (14%) fueron fallas. La tasa de reintervención en los sitios de fractura radiada fue del 13% frente al 10% en los sitios no irradiados. Cinco de los seis pacientes que se sometieron a reoperaciones por progresión del tumor local no habían recibido radioterapia en el sitio de la fractura, y ocho de diez seudoartrosis se habían tratado con radioterapia. Además, los cinco pacientes que tuvieron una fractura por estrés habían sido tratados con radioterapia.9 Todo esto confirma que las endoprótesis tienen una tasa de falla menor que los dispositivos osteosintéticos y que la radiación puede prevenir la progresión del tumor. |
| Mieloma múltiple | Los síntomas de presentación del mieloma múltiple suelen ser fatiga y dolor de huesos, pero los pacientes pueden tener otros síntomas asociados con insuficiencia orgánica, como insuficiencia renal, infección y anemia. Algunos pacientes pueden tener mieloma asintomático, donde las células anormales están presentes pero no los síntomas ni el daño orgánico. Los pacientes con MM pueden tener anemia normocítica, normocrómica secundaria a insuficiencia medular y aumento de la VSG. La hipercalcemia puede causar confusión, debilidad y letargo. Otros síntomas pueden incluir caquexia, compresión de la médula espinal e insuficiencia renal. Las infecciones bacterianas son comunes debido a la falta de producción normal de inmunoglobulinas.
La inmunoglobulina monoclonal (Ig G) se encuentra en la electroforesis sérica y en el análisis de orina. La inmunoglobulina (Ig) es la proteína que producen las células tumorales. Las subunidades de cadena ligera de inmunoglobulina se denominan proteínas de Bence Jones y están presentes en la orina. Las pruebas iniciales incluyen un ensayo cuantitativo de Ig, pruebas de función renal, calcio sérico y un nivel de microglobulina beta-2. Se requiere una biopsia de médula ósea para medir el porcentaje de células plasmáticas en la médula ósea. Más del 10% de células plasmáticas es una característica de diagnóstico del MM. El daño óseo es común y puede ser grave en el MM, y un gran porcentaje de pacientes desarrollará fracturas patológicas. Se recomienda una cuidadosa evaluación y monitoreo esquelético, así como una consulta cercana con un especialista en tumores ortopédicos. El agotamiento de los niveles de calcio / minerales óseos puede conducir a un riesgo de fractura grave, pero puede evitarse o minimizarse con el cuidado adecuado. Los tratamientos de conservación ósea deben instituirse lo antes posible. |
| Mieloma solitario | El dolor es el síntoma más común. Sin embargo, si el mieloma ocurre en la columna, generalmente se presentará con paraplejía de rápido desarrollo y deformidad gibosa debido al colapso vertebral. |
| Miositis osificante | La miositis osificante se presenta como un agrandamiento rápido y un dolor significativo una o dos semanas después de la lesión. El paciente tiene hinchazón y calor en el sitio, así como un aumento de la VSG y de la fosfatasa alcalina sérica. El cuadro clínico difiere del del osteosarcoma (que a menudo se encuentra en el diferencial porque los grupos de edad son los mismos) ya que el dolor de la miositis osificante disminuye a medida que pasa el tiempo y aumenta el dolor del osteosarcoma. Por lo general, los pacientes no presentan fiebre ni síntomas sistémicos. El dolor se localiza en el músculo afectado. |
| Osteosarcoma multifocal | |
| Mieloma múltiple | Los síntomas de presentación del mieloma múltiple suelen ser fatiga y dolor de huesos, pero los pacientes pueden tener otros síntomas asociados con insuficiencia orgánica, como insuficiencia renal, infección y anemia. Algunos pacientes pueden tener mieloma asintomático, donde las células anormales están presentes pero no los síntomas ni el daño orgánico. Los pacientes con MM pueden tener anemia normocítica, normocrómica secundaria a insuficiencia medular y aumento de la VSG. La hipercalcemia puede causar confusión, debilidad y letargo. Otros síntomas pueden incluir caquexia, compresión de la médula espinal e insuficiencia renal. Las infecciones bacterianas son comunes debido a la falta de producción normal de inmunoglobulinas.
La inmunoglobulina monoclonal (Ig G) se encuentra en la electroforesis sérica y en el análisis de orina. La inmunoglobulina (Ig) es la proteína que producen las células tumorales. Las subunidades de cadena ligera de inmunoglobulina se denominan proteínas de Bence Jones y están presentes en la orina. Las pruebas iniciales incluyen un ensayo cuantitativo de Ig, pruebas de función renal, calcio sérico y un nivel de microglobulina beta-2. Se requiere una biopsia de médula ósea para medir el porcentaje de células plasmáticas en la médula ósea. Más del 10% de células plasmáticas es una característica de diagnóstico del MM. El daño óseo es común y puede ser grave en el MM, y un gran porcentaje de pacientes desarrollará fracturas patológicas. Se recomienda una cuidadosa evaluación y monitoreo esquelético, así como una consulta cercana con un especialista en tumores ortopédicos. El agotamiento de los niveles de calcio / minerales óseos puede conducir a un riesgo de fractura grave, pero puede evitarse o minimizarse con el cuidado adecuado. Los tratamientos de conservación ósea deben instituirse lo antes posible. |
| Osteocondromas múltiples: pie y tobillo | Una sobrecarga en valgo del tobillo. |
| Osteocondromas múltiples: pie y tobillo | Una sobrecarga en valgo del tobillo. |
| Miositis osificante | La miositis osificante se presenta como un agrandamiento rápido y un dolor significativo una o dos semanas después de la lesión. El paciente tiene hinchazón y calor en el sitio, así como un aumento de la VSG y de la fosfatasa alcalina sérica. El cuadro clínico difiere del del osteosarcoma (que a menudo se encuentra en el diferencial porque los grupos de edad son los mismos) ya que el dolor de la miositis osificante disminuye a medida que pasa el tiempo y aumenta el dolor del osteosarcoma. Por lo general, los pacientes no presentan fiebre ni síntomas sistémicos. El dolor se localiza en el músculo afectado. |
| Neurofibroma | A menudo surgen como masas superficiales indoloras en la dermis. |
| Fibroma no osificante | Clínicamente, los fibromas no osificantes son asintomáticos y generalmente se descubren como un hallazgo incidental en la radiografía. En ocasiones, una lesión más grande se presenta como una fractura patológica. El escenario clásico es un niño que tiene una lesión menor en una competencia deportiva y se toma una radiografía simple en una sala de emergencias local. Se observa una lesión y se deriva al niño a un cirujano ortopédico para su evaluación. Por lo general, una historia clínica y un examen físico cuidadosos mostrarán que el dolor del niño está relacionado con la articulación cercana más que con la lesión, y que el patrón de los síntomas se ajusta al que se espera en un esguince o lesión por distensión. Si hubo dolor antes de la lesión que parece surgir de la lesión, el diagnóstico de NOF es altamente sospechoso. El síndrome de Jaffe-Campanacci es una constelación de síntomas que incluyen múltiples fibromas no osificantes, |
| No linfoma de Hodgkin | Puede presentarse como dolor o hinchazón local. Los pacientes generalmente sienten que gozan de buena salud de otra manera. |
| Fibroma no osificante | Clínicamente, los fibromas no osificantes son asintomáticos y generalmente se descubren como un hallazgo incidental en la radiografía. En ocasiones, una lesión más grande se presenta como una fractura patológica. El escenario clásico es un niño que tiene una lesión menor en una competencia deportiva y se toma una radiografía simple en una sala de emergencias local. Se observa una lesión y se deriva al niño a un cirujano ortopédico para su evaluación. Por lo general, una historia clínica y un examen físico cuidadosos mostrarán que el dolor del niño está relacionado con la articulación cercana más que con la lesión, y que el patrón de los síntomas se ajusta al que se espera en un esguince o lesión por distensión. Si hubo dolor antes de la lesión que parece surgir de la lesión, el diagnóstico de NOF es altamente sospechoso. El síndrome de Jaffe-Campanacci es una constelación de síntomas que incluyen múltiples fibromas no osificantes, |
| Síndrome de Ollier | El niño tiene un crecimiento anormal, acortamiento y deformidad de la extremidad afectada. |
| Osteoblastoma | Los síntomas comunes son dolor de larga duración, hinchazón y sensibilidad. Los tumores de la columna vertebral pueden causar escoliosis y síntomas neurológicos. La lesión puede presentarse clínicamente con síntomas mielopáticos y / o radiculares. |
| Osteoblastoma: pie y tobillo |
| Osteoblastoma: pie y tobillo | |
| Osteocondroma | Clínicamente, los osteocondromas se presentan con dolor debido a irritación mecánica o una masa indolora. Puede ocurrir una fractura a través del tallo de la lesión que también causa dolor. |
| Osteocondromatosis | Aproximadamente el 50% de los niños afectados tienen lesiones identificables a la edad de 5 años. Los pacientes presentan desfiguración o dolor inducido por la presión sobre las estructuras de tejidos blandos circundantes. Pueden tener baja estatura, discrepancias en la longitud de las extremidades, angulación en valgo de la rodilla y el tobillo, arqueamiento del radio con desviación cubital de la muñeca y subluxación de la articulación radiocapitellar. El cúbito y el peroné se ven más afectados que el radio y la tibia. Por esta razón, el crecimiento diferencial de los huesos emparejados puede dar lugar a deformidades angulares del antebrazo, la muñeca, la pierna y el tobillo. Además, los grupos de lesiones en los extremos metafisarios de los huesos cerca de la articulación pueden provocar pérdida de movimiento, displasia de cadera, subluxación de la articulación, compresión nerviosa, pseudoaneurisma vascular, compresión de la médula y dolor. |
| Osteocondroma | Clínicamente, los osteocondromas se presentan con dolor debido a irritación mecánica o una masa indolora. Puede ocurrir una fractura a través del tallo de la lesión que también causa dolor. |
| Osteocondroma – de Pie y Tobillo | |
| Osteocondromatosis | Aproximadamente el 50% de los niños afectados tienen lesiones identificables a la edad de 5 años. Los pacientes presentan desfiguración o dolor inducido por la presión sobre las estructuras de tejidos blandos circundantes. Pueden tener baja estatura, discrepancias en la longitud de las extremidades, angulación en valgo de la rodilla y el tobillo, arqueamiento del radio con desviación cubital de la muñeca y subluxación de la articulación radiocapitellar. El cúbito y el peroné se ven más afectados que el radio y la tibia. Por esta razón, el crecimiento diferencial de los huesos emparejados puede dar lugar a deformidades angulares del antebrazo, la muñeca, la pierna y el tobillo. Además, los grupos de lesiones en los extremos metafisarios de los huesos cerca de la articulación pueden provocar pérdida de movimiento, displasia de cadera, subluxación de la articulación, compresión nerviosa, pseudoaneurisma vascular, compresión de la médula y dolor. |
| Displasia osteofibrosa | El tumor se presenta clínicamente como una masa que aumenta de tamaño e indolora. |
| Osteoma osteoide | El osteoma osteoide tiene un cuadro clínico distintivo de dolor sordo que empeora por la noche y desaparece entre 20 y 30 minutos después del tratamiento con medicamentos antiinflamatorios no esteroideos. Puede haber dolor articular con una lesión periarticular y puede ocurrir sinovitis secundaria a una lesión intraarticular. Los síntomas locales pueden incluir un aumento de la temperatura de la piel, aumento de la sudoración y sensibilidad. Las lesiones epifisarias pueden causar un crecimiento anormal. |
| Osteoide osteoide: pie y tobillo | Los pacientes se presentan con dolor e hinchazón persistentes, que no están relacionados con la actividad. El dolor puede ser más intenso por la noche. |
| Osteoma | Los osteomas son lesiones de crecimiento lento que normalmente son completamente asintomáticas. Solo se presentan si su ubicación dentro de la región de la cabeza y el cuello está causando problemas con la respiración, la visión o la audición. |
| Osteoma | Los osteomas son lesiones de crecimiento lento que normalmente son completamente asintomáticas. Solo se presentan si su ubicación dentro de la región de la cabeza y el cuello está causando problemas con la respiración, la visión o la audición. |
| Osteoma osteoide | El osteoma osteoide tiene un cuadro clínico distintivo de dolor sordo que empeora por la noche y desaparece entre 20 y 30 minutos después del tratamiento con medicamentos antiinflamatorios no esteroideos. Puede haber dolor articular con una lesión periarticular y puede ocurrir sinovitis secundaria a una lesión intraarticular. Los síntomas locales pueden incluir un aumento de la temperatura de la piel, aumento de la sudoración y sensibilidad. Las lesiones epifisarias pueden causar un crecimiento anormal. |
| Osteoide osteoide: pie y tobillo | Los pacientes se presentan con dolor e hinchazón persistentes, que no están relacionados con la actividad. El dolor puede ser más intenso por la noche. |
| Osteomielitis | En los niños, la diseminación hematógena de la infección se presenta como fiebre alta, malestar, dolor local e hinchazón. El cuadro clínico puede ser ambiguo y puede surgir la preocupación por la posibilidad de un tumor. El microorganismo más común en pacientes mayores de tres años es el estafilococo coagulasa positivo. En adultos, la presentación clínica puede ser bastante engañosa, ya que a menudo faltan fiebre, recuento elevado de leucocitos y antecedentes de una posible fuente de infección. Los usuarios de drogas intravenosas a menudo tienen organismos inusuales como pseudomonas. Los pacientes mayores pueden tener bacterias gramnegativas en la columna secundaria a organismos que se originan como una infección del tracto urinario y viajan a través del plexo de Batson. La osteomielitis espinal puede presentarse como dolor de espalda con hemocultivos negativos. |
| Osteomielitis | En los niños, la diseminación hematógena de la infección se presenta como fiebre alta, malestar, dolor local e hinchazón. El cuadro clínico puede ser ambiguo y puede surgir la preocupación por la posibilidad de un tumor. El microorganismo más común en pacientes mayores de tres años es el estafilococo coagulasa positivo. En adultos, la presentación clínica puede ser bastante engañosa, ya que a menudo faltan fiebre, recuento elevado de leucocitos y antecedentes de una posible fuente de infección. Los usuarios de drogas intravenosas a menudo tienen organismos inusuales como pseudomonas. Los pacientes mayores pueden tener bacterias gramnegativas en la columna secundaria a organismos que se originan como una infección del tracto urinario y viajan a través del plexo de Batson. La osteomielitis espinal puede presentarse como dolor de espalda con hemocultivos negativos. |
| Osteopoikilosis | Los pacientes están asintomáticos y los exámenes de laboratorio son normales. Se ha informado de la aparición de osteosarcoma en el hueso afectado. Las condiciones asociadas incluyen: dermatofibrosis lenticularis diseminada, esclerodermia, sindactilia, enanismo, anomalías endocrinas, melorreostosis y paladar hendido. La mayoría de los pacientes no presentan otros hallazgos. |
| Osteopoikilosis | Los pacientes están asintomáticos y los exámenes de laboratorio son normales. Se ha informado de la aparición de osteosarcoma en el hueso afectado. Las condiciones asociadas incluyen: dermatofibrosis lenticularis diseminada, esclerodermia, sindactilia, enanismo, anomalías endocrinas, melorreostosis y paladar hendido. La mayoría de los pacientes no presentan otros hallazgos. |
| Osteosarcoma – convencional | La presentación más común es dolor y una masa, que ocurre cerca de una articulación. El dolor puede acompañar inicialmente a la actividad, pero gradualmente se vuelve más constante y puede ser intenso por la noche. De lo contrario, el paciente puede tener pocos síntomas. |
| Osteosarcoma – Pie y tobillo | El paciente presenta un dolor insidioso y, finalmente, una masa. Inicialmente, el dolor puede ser intermitente y estar relacionado con una lesión menor o una actividad de ejercicio y, por lo tanto, el problema se diagnostica erróneamente como un esguince o distensión común.
El dolor se volverá persistente con el tiempo y comenzará a ocurrir por la noche. |
| Osteosarcoma – Pie y tobillo | El paciente presenta un dolor insidioso y, finalmente, una masa. Inicialmente, el dolor puede ser intermitente y estar relacionado con una lesión menor o una actividad de ejercicio y, por lo tanto, el problema se diagnostica erróneamente como un esguince o distensión común.
El dolor se volverá persistente con el tiempo y comenzará a ocurrir por la noche. |
| Osteosarcoma – convencional | La presentación más común es dolor y una masa, que ocurre cerca de una articulación. El dolor puede acompañar inicialmente a la actividad, pero gradualmente se vuelve más constante y puede ser intenso por la noche. De lo contrario, el paciente puede tener pocos síntomas. |
| Osteosarcoma de superficie de alto grado | |
| Osteosarcoma de células pequeñas | Similar a los del osteosarcoma convencional. |
| Osteosarcoma perióstico | El paciente presenta dolor local y una masa, que puede estar en la diáfisis y la cortical anterior del hueso, especialmente cuando el tumor afecta la tibia. |
| Enfermedad de Paget | El sarcoma de Paget generalmente se presenta como un dolor nuevo y progresivo en un paciente con enfermedad de Paget de larga duración. Otros síntomas pueden incluir hinchazón de tejidos blandos o fracturas patológicas. La fosfatasa alcalina sérica que se eleva con la enfermedad de Paget puede aumentar aún más con la aparición del sarcoma. |
| Osteosarcoma parosteal | El tumor se presenta con una masa de crecimiento lento y dolor leve. Ocasionalmente, los pacientes no sienten ningún dolor. Las ubicaciones más comunes son el fémur distal cerca de la parte posterior de la rodilla y el húmero proximal cerca del hombro. |
| Condroma perióstico | Los síntomas están presentes durante 1 a 5 años. Los pacientes se quejan de una masa o hinchazón dolorosa. |
| Condroma perióstico – pie y tobillo | La lesión se presenta con dolor e hinchazón. |
| Osteosarcoma perióstico | El paciente presenta dolor local y una masa, que puede estar en la diáfisis y la cortical anterior del hueso, especialmente cuando el tumor afecta la tibia. |
| Periostitis florida reactiva | El examen revela una masa sensible con leve calor circundante y eritema, pero sin fiebre ni adenopatía. Los estudios de laboratorio suelen mostrar un recuento y diferencial de glóbulos blancos normales y una tasa de sedementación levemente elevada. Los electrolitos y otros valores de laboratorio no se ven afectados. Las radiografías simples muestran que el tumor está adyacente al hueso en lugar de surgir de él. Se observa inflamación de los tejidos blandos y una marcada reacción perióstica. Puede haber formación de hueso nuevo en los tejidos blandos y adelgazamiento cortical sutil. En casos raros hay erosión local de la corteza. Las gammagrafías óseas con tecnecio-99 muestran un foco solitario de captación intensa. Las exploraciones por resonancia magnética muestran una masa y anomalías de señal llamativas en los tejidos blandos, así como en la médula ósea cercana, que son relativamente inespecíficas y son compatibles con una infección, un traumatismo o un tumor. |
| Sinovitis vellonodular pigmentada | Se presenta como una articulación indolora o levemente dolorosa con hinchazón. |
| Sinovitis vellonodular pigmentada: pie y tobillo | La lesión se presenta más comúnmente como una masa nodular localizada en o adyacente a una articulación o vaina tendinosa. La forma nodular ocurre alrededor de las articulaciones y tendones de las manos y los pies. La forma difusa generalmente involucra articulaciones más grandes y puede involucrar varias articulaciones adyacentes. |
| Fibroma plantar – pie y tobillo | La mayoría de los pacientes son asintomáticos, pero algunos tienen dolor relacionado con la actividad. Entre un tercio y la mitad de los pacientes tienen nódulos bilaterales. El dolor se presenta con las actividades que soportan peso. Cuando las lesiones son lo suficientemente grandes como para presionar los nervios plantares, puede haber entumecimiento o disestesia en las porciones distales del pie. En un paciente pediátrico, una gran lesión provocó la contractura de los tendones flexores del dedo gordo y la pérdida de la extensión de los dedos inferiores. |
| Polimórfica Fibroóseo Lesión del Hueso | Este tumor se presenta con dolor. |
| Post – Sarcoma de Paget | El sarcoma de Paget generalmente se presenta como un dolor nuevo y progresivo en un paciente con enfermedad de Paget de larga duración. Otros síntomas pueden incluir hinchazón de tejidos blandos o fracturas patológicas. La fosfatasa alcalina sérica que se eleva con la enfermedad de Paget puede aumentar aún más con la aparición del sarcoma. Los pacientes a menudo se quejan de dolor antes de que el tumor pueda verse fácilmente en una radiografía simple y el diagnóstico a menudo puede retrasarse. Con el tiempo, se hace evidente la destrucción cortical y una masa tumoral mal definida que se extiende hacia los tejidos blandos con un trasfondo de enfermedad de Paget. Si se encuentra un sarcoma de Paget, se debe examinar todo el cuerpo, ya que el tumor a menudo surge en más de un sitio. |
| Post – Sarcoma de Paget | El sarcoma de Paget generalmente se presenta como un dolor nuevo y progresivo en un paciente con enfermedad de Paget de larga duración. Otros síntomas pueden incluir hinchazón de tejidos blandos o fracturas patológicas. La fosfatasa alcalina sérica que se eleva con la enfermedad de Paget puede aumentar aún más con la aparición del sarcoma. Los pacientes a menudo se quejan de dolor antes de que el tumor pueda verse fácilmente en una radiografía simple y el diagnóstico a menudo puede retrasarse. Con el tiempo, se hace evidente la destrucción cortical y una masa tumoral mal definida que se extiende hacia los tejidos blandos con un trasfondo de enfermedad de Paget. Si se encuentra un sarcoma de Paget, se debe examinar todo el cuerpo, ya que el tumor a menudo surge en más de un sitio. |
| Osteosarcoma posradiación – sarcoma posradiación | Los síntomas pueden pasar desapercibidos durante bastante tiempo o ser malinterpretados por el paciente y el médico, y luego se produce un inicio agudo de dolor y / o hinchazón constante y progresivo. El dolor empeora por la noche y generalmente no se alivia con aspirina o medicamentos no esteroides. Otros síntomas incluyen el desarrollo de una masa y / o una fractura patológica cerca del área de irradiación con dolor a la palpación y / o sangrado. |
| golpe del corredor – Pie y tobillo | La masa generalmente se presenta después de un cambio menor en el programa de entrenamiento o una lesión insignificante. La lesión aumenta de tamaño con la carrera y las actividades y disminuye con el reposo y la elevación. El dolor suele ser lo suficientemente leve como para permitir la continuación del programa en ejecución, lo que a su vez conduce a la persistencia de la lesión. |
| Quiste óseo aneurismático | Dolor que aumenta gradualmente, una masa o con una fractura patológica a través de la lesión. En unos pocos casos se ha informado de un rápido aumento del tamaño de la lesión. |
| Quiste óseo unicameral | La mayoría de los CBU son asintomáticos y solo están presentes cuando se produce una fractura patológica. |
| Quiste óseo unicameral – pie y tobillo | Se presenta como un hallazgo incidental o con dolor leve durante la práctica de deportes o la carrera. |
| golpe del corredor – Pie y tobillo | La masa generalmente se presenta después de un cambio menor en el programa de entrenamiento o una lesión insignificante. La lesión aumenta de tamaño con la carrera y las actividades y disminuye con el reposo y la elevación. El dolor suele ser lo suficientemente leve como para permitir la continuación del programa en ejecución, lo que a su vez conduce a la persistencia de la lesión. |
| Sarcoma de Ewing | La presentación clínica del sarcoma de Ewing incluye dolor e hinchazón de semanas o meses de duración. A veces se observan eritema y calor en el área local. La osteomielitis es a menudo el diagnóstico inicial basado en fiebres intermitentes, leucocitosis, anemia y aumento de la VSG. |
| Sarcoma granulocítico en hueso | La mayoría de los sarcomas granulocíticos óseos se presentan con dolor esquelético atribuible a la ubicación de la lesión. Este tumor tiene tres presentaciones clínicas características: una, en un individuo sin enfermedad conocida donde es un presagio de leucemia mielógena aguda; dos, en un individuo con un trastorno mieloproliferativo ya conocido. La evaluación posterior de la biopsia de sangre periférica y médula ósea en este caso demostró que se trataba de un sarcoma granulocítico sin evidencia sistémica de leucemia mielógena aguda. La aparición del tumor en un individuo previamente sano presenta un importante desafío diagnóstico y el 75% de estos casos suelen ser mal diagnosticados. |
| Schwannoma – neurilemoma – pie y tobillo | Los tumores son de crecimiento lento y la transformación maligna es rara (2). La mayoría de las lesiones son asintomáticas. El tumor solitario típico se presenta como una masa indolora de crecimiento lento que puede haber estado presente durante 1 a 2 años o más. El descubrimiento de un schwannoma debería desencadenar una búsqueda cuidadosa de otros. |
| Schwannoma de hueso | La mayoría de las lesiones son asintomáticas. |
| Schwannoma de hueso | La mayoría de las lesiones son asintomáticas. |
| Condrosarcoma secundario | |
| Síndrome de Jaffe-Campanacci | Las lesiones cutáneas son parches café con leche, que son de color marrón a marrón oscuro y generalmente se encuentran en el tronco. No todos los pacientes con este síndrome tienen parches cutáneos. La mayoría de los parches tenían un borde liso. También se pueden encontrar parches de café con leche con bordes complejos. Las características cutáneas de la neurofibromatosis (NF), como angiomas y neurofibromas, también pueden estar presentes. Puede haber pecas axilares. Hallazgos recientes sugieren que JCS puede ser una forma de NF. Los pacientes con sospecha de JCS deben someterse a pruebas de detección de NF. |
| El síndrome de Mafucci | La enfermedad puede estar localizada o diseminada. La mayoría de los pacientes se presentan con dolor. Pueden aparecer otros tumores. El número de pacientes que desarrollan sarcomas varía según los informes. Los pacientes con lesiones extensas tienen un mayor riesgo. Aproximadamente el 15% desarrolla condrosarcoma y un tercio tiene otros sarcomas, incluidos sarcomas vasculares y fibrosos. La tasa de condrosarcoma es menor que la observada en la enfermedad de Ollier. Pueden aparecer otros tumores benignos y malignos, como adenoma hipofisario, glioma, fibromas y pólipos uterinos, tumores ováricos mesenquimales, adenoma cortical suprarrenal, fibrosarcoma y carcinoma de páncreas. |
| Síndrome de McCune-Albright (MAS) | La mutación del gen GS en el cromosoma 20q13 ocurre temprano en el desarrollo y da como resultado un mosaico de células anormales y mutadas. Las manifestaciones de MAS en cada individuo dependen de la extensión y distribución de las células anormales. La activación anormal y prolongada de múltiples glándulas endocrinas periféricas ocurre incluso cuando las hormonas hipofisarias estimulantes necesarias pueden estar ausentes. En las mujeres puede ocurrir pubertad precoz, con inicio del desarrollo de las mamas, vello púbico y el inicio de la menstruación ya en los primeros meses de vida. Otras manifestaciones incluyen acromegalia, hipertiroidismo, hiperprolactinemia y otras.
Los huesos frecuentemente afectados incluyen el fémur, la tibia, el esqueleto facial y las costillas. La fragilidad ósea y las fracturas asociadas son comunes, y los huesos que soportan peso pueden sufrir múltiples fracturas. En los fémures proximales, múltiples microfracturas corticales sucesivas pueden dar como resultado un arqueamiento característico del extremo proximal del hueso en una deformidad en “curva de pastor”. |
| Sinovitis vellonodular pigmentada | Se presenta como una articulación indolora o levemente dolorosa con hinchazón. |
| Sinovitis vellonodular pigmentada: pie y tobillo | La lesión se presenta más comúnmente como una masa nodular localizada en o adyacente a una articulación o vaina tendinosa. La forma nodular ocurre alrededor de las articulaciones y tendones de las manos y los pies. La forma difusa generalmente involucra articulaciones más grandes y puede involucrar varias articulaciones adyacentes. |
| Osteosarcoma de células pequeñas | Similar a los del osteosarcoma convencional. |
| Tumor fibroso solitario de hueso | Suele presentarse con dolor y lesiones recurrentes. |
| Mieloma solitario | El dolor es el síntoma más común. Sin embargo, si el mieloma ocurre en la columna, generalmente se presentará con paraplejía de rápido desarrollo y deformidad gibosa debido al colapso vertebral. |
| Quiste subcondral | Inicio y progresión gradual del dolor en la articulación. |
| Quiste subcondral – pie y tobillo | |
| Quiste subcondral | Inicio y progresión gradual del dolor en la articulación. |
| Condromatosis sinovial | La condromatosis sinovial se presenta como la aparición gradual de dolor y rigidez monoarticular. Si se permite que continúen, los síntomas lentos y progresivos pueden resultar en una disminución del rango de movimiento, derrames, crepitaciones y eventual bloqueo de la articulación. Esta condición también se ha descrito en la membrana sinovial, tejidos blandos y bursas. La condromatosis sinovial secundaria puede estar presente después de una osteoartritis de larga duración. Existen varias etiologías posibles de la condromatosis sinovial. El trauma está respaldado por el hecho de que el proceso ocurre principalmente en las articulaciones dominantes de las extremidades superiores y en las que soportan peso. La infección es otro posible culpable. Otra teoría postula que el cartílago desprendido hacia la articulación es absorbido por la membrana sinovial. |
| Condromatosis sinovial – pie y tobillo | El inicio se describe como insidioso y se produce durante meses o años [2]. Iossifidis et al describieron una presentación clínica insidiosa e inespecífica en su caso de condromatosis sinovial del tobillo [6]. El diagnóstico de condromatosis sinovial a menudo se realiza después de una anamnesis, un examen físico y un examen radiográficos completos. Los pacientes pueden informar dolor e hinchazón dentro de una articulación. Esto se agrava habitualmente con la actividad física. Por lo general, el paciente también puede referir dolor, rango de movimiento reducido, nódulos palpables, bloqueo o chasquido de la articulación [7,11]. Estas lesiones pueden volverse sintomáticas después de la compresión mecánica o la irritación de los tejidos blandos, los nervios o la transformación maligna. En casos raros, se pueden formar bursas reactivas sobre osteocondromas. Éstos pueden ser otra fuente de dolor, pero también pueden simular condrosarcoma [14]. Por el contrario, las personas pueden no tener signos o síntomas y es un hallazgo incidental secundario a otra queja. Según Milgram, esto está relacionado con el estadio de la lesión [15]. |
| Sarcoma sinovial: pie y tobillo | La presentación del sarcoma sinovial es variable y puede simular un proceso benigno como el quiste ganglionar. El paciente puede tener una masa que ha estado presente durante meses, años o incluso décadas, con crecimiento lento y poco o ningún síntoma. Es posible que haya habido un crecimiento rápido reciente de una lesión que ha estado presente durante años sin cambios aparentes. Por el contrario, algunos de estos sarcomas pueden ser muy dolorosos desde el principio. La duración media de los síntomas antes del diagnóstico es de 21 meses. |
| Osteosarcoma telangectásico | Se presenta más comúnmente como una fractura patológica que el osteosarcoma convencional, pero tiene una epidemiología similar. |
| Prueba Tumor Uno / Prueba Tumor Uno | |
| Tumor de células gigantes | La mayoría de los pacientes se presentan con dolor de progresión lenta, con o sin una masa. Los síntomas surgen cuando la lesión comienza a destruir la corteza e irrita el periostio o cuando el debilitamiento del hueso causado por el tumor provoca dolor por fractura patológica inminente. Algunos tumores de células gigantes se presentan con una fractura patológica. |
| Tumor de células gigantes: pie y tobillo | El paciente típico tiene antecedentes de dolor que aumenta gradualmente. En el pie puede aparecer una masa debido a la limitación de los tejidos blandos. El dolor por fractura patológica o microfractura puede hacer que el paciente busque tratamiento. |
| Tumor fibroso solitario de hueso | Suele presentarse con dolor y lesiones recurrentes. |
| Tumor glómico | Clínicamente, los tumores glómicos se caracterizan por una tríada de sensibilidad al frío, sensibilidad localizada y dolor intermitente intenso. El dolor puede ser insoportable y se describe como ardor o estallido. La causa exacta del dolor no se comprende completamente, pero se han identificado fibras nerviosas que contienen la sustancia neurotransmisora del dolor P en el tumor. |
| Mímicos de tumores | Gota La gota es una enfermedad causada por altos niveles de ácido úrico en el torrente sanguíneo. Los cristales de ácido úrico se depositan en el cartílago articular de las articulaciones provocando rigidez y episodios dolorosos de artritis aguda. La gota puede presentarse en un paciente atípico o en una ubicación atípica del pie, de modo que el médico no incluya la gota en el diferencial. En estos casos inusuales, el diagnóstico puede pasarse por alto incluso cuando el cuadro clínico es típico de la gota.Fractura por estrés Algunos pacientes presentarán dolor y una fractura por estrés temprana sin antecedentes de actividad prolongada o caminar. Es posible que los niños no puedan dar una historia que sea suficiente para que el médico considere la fractura por estrés en el diferencial. Las radiografías simples también pueden malinterpretarse.La PVNS sinovitis vellonodular pigmentada puede causar lesiones quísticas óseas que imitan un proceso neoplásico. |
| Mímicos de tumores | Gota La gota es una enfermedad causada por altos niveles de ácido úrico en el torrente sanguíneo. Los cristales de ácido úrico se depositan en el cartílago articular de las articulaciones provocando rigidez y episodios dolorosos de artritis aguda. La gota puede presentarse en un paciente atípico o en una ubicación atípica del pie, de modo que el médico no incluya la gota en el diferencial. En estos casos inusuales, el diagnóstico puede pasarse por alto incluso cuando el cuadro clínico es típico de la gota.Fractura por estrés Algunos pacientes presentarán dolor y una fractura por estrés temprana sin antecedentes de actividad prolongada o caminar. Es posible que los niños no puedan dar una historia que sea suficiente para que el médico considere la fractura por estrés en el diferencial. Las radiografías simples también pueden malinterpretarse.La PVNS sinovitis vellonodular pigmentada puede causar lesiones quísticas óseas que imitan un proceso neoplásico. |
| Mímicos de tumores: gota | |
| Imitadores de tumores – Fractura por estrés | |
| Calcinosis tumoral | |
| Quiste óseo unicameral | La mayoría de los CBU son asintomáticos y solo están presentes cuando se produce una fractura patológica. |
| Quiste óseo unicameral – pie y tobillo | Se presenta como un hallazgo incidental o con dolor leve durante la práctica de deportes o la carrera. |
| Condroma yuxtacortical | Dolor leve o síntomas que surgen de la articulación cercana. |